|
ГЕТЕРОГЕННЫЙ КАТАЛИЗ (контактный катализ), изменение скорости
хим. р-ции при воздействии катализаторов, образующих самостоят. фазу и
отделенных от реагирующих в-в границей раздела. наиб. распространен случай,
когда твердый кат. (контакт) ускоряет р-цию между газообразными реагентами
или р-цию в р-ре. Каталитич. р-ция протекает обычно на пов-сти твердого
кат. и обусловлена активацией молекул реагентов при взаимод. с пов-стью.
Поэтому для осуществления Г. к. необходима адсорбция компонентов реакц.
смеси из объемной фазы на пов-сти катализатора.
В техн. Г. к. св-во катализатора ускорять р-цию обычно определяют как
выход продукта в единицу времени, отнесенный к единице объема или массы
катализатора. В теоретич. исследованиях скорость v гетерогенно-каталитич.
р-ций относят к единице пов-сти катализатора и наз. удельной каталитич.
активностью; ее размерность - моль х х с-1 м-2(см.
Активность катализатора). Если все активные центры пов-сти однородны
и равнодоступны молекулам реагирующих в-в, v пропорциональна пов-сти
S:
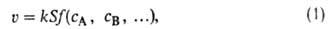
где f- ф-ция концентраций сA, сB, ... реагентов
и продуктов р-ции в объемной фазе, k-константа скорости, отнесенная
к единице пов-сти катализатора. Соотношение (1) м. б. неприменимо, если
гетерогенно-каталитич. р-ция осложнена диффузией реагирующих молекул, в
случае неоднородной пов-сти и др. (подробнее см. Каталитических реакций
кинетика).
Развитие эксперим. техники сделало возможным в ряде случаев относить
скорость гетерогенно-каталитич. р-ции к единичному активному центру пов-сти.
Применение сверхвысокого вакуума (~10-8Па) позволило получить
атомарно чистые пов-сти металлов (свободные от адсорбиров. частиц), на
к-рых все атомы (их число Nc ~ 1019 м-2)
являются активными центрами. Число молекул, подвергающихся превращениям
на одном активном центре в секунду, наз. числом оборотов р-ции tn.
Скорость р-ции связана с tn соотношением:

Для многих р-ций tn составляет от 10-2
до 102 с -1 однако возможны как существенно меньшие,
так и большие значения. Напр., в случае окисления NH3 на пром.
катализаторе tn достигает 5*104 с-1,
а для той же р-ции на монокристалле Pt при низком давлении -10-6
с-1.
Число активных центров на катализаторах-оксидах металлов оценить труднее.
Спектральные методы (оптическая и ИК-спектроскопия, ЭПР и др.) позволяют
следить за изменением структуры пов-сти этих в-в в условиях Г. к. и соотносить
эти изменения с изменениями скорости р-ции; отсюда можно оценить поверхностную
концентрацию активных центров. Оказалось, что для большинства оксидных
кат. число активных центров меньше, чем для металлов, и составляет 1014-1018
м-2.
Константы скорости каталитич. р-ции изменяются с т-рой Г в соответствии
с ур-нием Аррениуса: k = = k0ехр( — E/RT), где k0-предэкспоненц.
множитель, Е-энергия активации, R- газовая постоянная. Величина
k0 м. б. вычислена методами активированного комплекса
теории, однако сопоставление расчетных значений с экспериментальными
возможно лишь при условии, если точно известно число активных центров на
пов-сти реального катализатора. Значения Е определяются из эксперим.
зависимости k от Т, обычно они составляют 10-150 кДж/моль.
Катализаторы Г. к. обладают высокой селективностью, т. е. относит. способностью
ускорять одну из неск. одновременно протекающих р-ций. Часто на одном и
том же катализаторе могут протекать одновременно неск. последовательных
и параллельных р-ций. Напр., каталитич. окисление углеводородов может протекать
последовательно-сначала до получения ценных кислородсодержащих соед., затем
до полного их окисления с образованием СО2 и Н2О.
Наряду с этим может протекать и параллельное окисление исходного в-ва непосредственно
до СО2 и Н2О, без выхода ценных промежут. продуктов
мягкого окисления в газовую фазу. В простейшем случае мягкого окисления
углеводорода СnНm+2, до продукта СnНmО
(напр., при получении этиленоксида С2Н2О из этилена
С2Н4) процесс м. б. описан схематически след, образом:
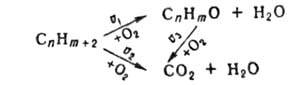
Селективность s катализатора определяется как отношение скорости накопления
целевого продукта к сумме скоростей всех р-ций, протекающих с участием
исходных в-в:

Стадия гетерогенного катализа - Сложная гетерогенно-каталитич.
р-ция протекает через последовательность стадий, традиционно считавшихся
элементарными: подвод реагентов из объемной фазы к пов-сти катализатора
(диффузия), адсорбция, хим. превращение на пов-сти (собственно катализ),
десорбция продуктов, их отвод от пов-сти катализатора (диффузия). Исходными
в-вами, реагирующими в элементарных р-циях, по определению, являются промежут.
в-ва, к-рые образуются в предшествующих стадиях и не м. б. выделены вместе
с реагирующими в-вами или продуктами р-ции. Предполагалось, что в Г. к.
такими в-вами являются поверхностные соед. катализатора с реагентами, а
стадии, в к-рых эти соед. образуются или распадаются,-элементарные. Развитие
эксперим. методов исследования позволило установить, что в нек-рых из стадий,
предполагавшихся ранее элементарными, можно выделить свои элементарные
процессы, в к-рых промежут. в-ва не образуются, а изменение состояния частицы
(атома, молекулы) связано с преодолением одного потенциального барьера.
Рассмотрим влияние каждой из стадий на кинетич. закономерности Г. к.
Диффузия. Если катализатор представляет собой пористые частицы (зерна),
в общем случае могут иметь место след. диффузионные процессы: 1) перенос
реагирующих в-в из объема газовой или жидкой фазы к внеш. пов-сти зерна
катализатора (внешняя диффузия); 2) перенос частиц в порах внутри зерна
(внутренняя диффузия); 3) и 4) обратные процессы - перенос частиц продуктов
р-ции изнутри пор к внеш. пов-сти зерен и отвод их от внеш. пов-сти зерен
в пространство между ними (соотв. внутр. и внеш. диффузия). Все эти процессы
обусловлены градиентом концентраций компонентов реакц. смеси в пределах
одной фазы или у границы раздела фаз. В зависимости от условий осуществления
процесса (т-ры, давления, св-в катализатора) различают кинетич. область
протекания Г. к., в к-рой все диффузионные процессы происходят значительно
быстрее, чем хим. р-ции, и диффузионные области (соотв. внешне- и внутренне-диффузионную),
в к-рых суммарный процесс тормозится более медленным, чем хим. р-ция, переносом
частиц. Поскольку с ростом т-ры и концентрации реагентов скорость хим.
р-ции увеличивается быстрее, чем диффузия, диффузионное торможение в Г.
к. происходит при высоких т-рах и давлениях, а также на высокодисперсных
катализаторах. Если ур-ние р-ции в кинетич. области имеет вид:

где n-порядок р-ции, с-концентрация реагирующего в-ва в объемной фазе,
то с повышением т-ры и переходом во внутренне-диффузионную область скорость
р-ции описывается ур-нием:
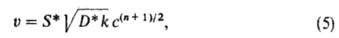
где S*-внеш. пов-сть зерна катализатора, D*- эффективный коэф. диффузии
в пористой среде. Поскольку в ур-ние входит константа скорости k в степени
1/2, измеряемая энергия активации р-ции во внутренне-диффузионной
области уменьшается вдвое по сравнению с ее значением в кинетич. области,
а порядок р-ции по реагенту, диффузия к-рого лимитирует суммарную скорость
процесса, изменяется до значения (п + 1)/2. При еще более высоких
т-рах, когда процесс переходит во внешне-диффузионную область, температурная
зависимость кажущейся константы скорости определяется температурной зависимостью
коэф. диффузии (см. рис. 1). Подробнее см. в ст. Макрокинетика.
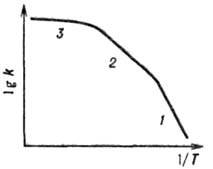
Рис. 1. Зависимость константы скорости k гетерогенно-каталитич. р-ции
от т-ры Т в кинетической (1), внутренне-диффузионной (2) и внешне-диффузионной
(3) областях протекания.
Адсорбция. Согласно общепринятым представлениям, основное значение в
Г. к. имеет хемосорбция, при к-рой адсорбируемые частицы химически связаны
с поверхностными атомами твердого тела. Для участия в послед. каталитич.
превращениях хемосорбиров. частица (атом, молекула) должна быть активирована,
т. е. переведена в более реакционноспособное (по сравнению с исходным)
состояние. Этот процесс может требовать затраты энергии (преодоления энергетич.
барьера), протекать медленно и оказаться лимитирующей стадией Г. к. Часто
медленную хемосорбцию в Г. к. наз. активированной адсорбцией. Примером
процесса, лимитируемого активиров. адсорбцией, является синтез NH3
на железном кат., скорость к-рого определяется адсорбцией N2
на пов-сти Fe.
Энергия связи хемосорбиров. частицы с пов-стью катализатора не должна
быть ни слишком большой, ни слишком малой. Так, изменение скорости окисления
водорода на пов-сти разл. оксидов металлов имеет максимум, соответствующий
оптимальной для катализа теплоте адсорбции Qs кислорода
(рис. 2). Слабая связь (напр., при физ. адсорбции) не приводит к активации
адсорбиров. частицы и образованию более реакционноспособного состояния,
а слишком прочная связь затрудняет дальнейшие превращ. (повышает энергию
активации послед. стадии).
Причины активиров. адсорбции при Г. к. могут быть различными. Больших
энергетич. затрат может требовать перестройка поверхностной структуры катализатора.
Напр., при адсорбции Н2 или СО на грани (100) кубич. монокристаяла
Pt гексагональная структура перестраивается в квадратную. Элементарными
процессами активации м. б. также перенос электрона от катализатора к хемосорбиров.
молекулам с образованием ионов или своб. радикалов, подвод энергии к адсорбиров.
частице с образованием колебательно- или электронно-возбужденных молекул,
взаимная ориентация атомов или атомных групп хемосорбиров. молекул, благоприятная
для послед. образования реакционноспособных комплексов, напр.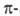 аллильных
комплексов при адсорбции олефинов, и др. аллильных
комплексов при адсорбции олефинов, и др.
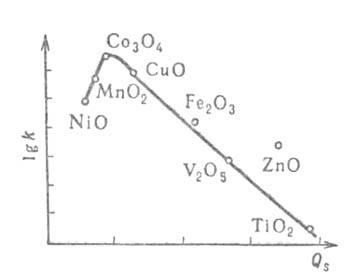
Рис. 2. Зависимость константы скорости k окисления Н2
от теплоты адсорбции Q, кислорода (энергии связи металл -кислород).
Во мн. процессах Г. к. адсорбция реагирующих в-в на пов-сти катализатора
происходит через образование т. наз. предсорбционного состояния, или прекурсора,
к-рое далее либо участвует в катализе, либо препятствует ему. Так, при
окислении СО на МnО2 образуется предсорбц. состояние МnО2(СО),
к-рое далее может участвовать в образовании продукта СО2 либо
ведет к прочной хемосорбции с образованием поверхностного карбоната МnСО3,
к-рый отравляет катализатор:
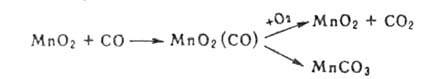
Др. пример-образование на пов-сти слабо связанного прекурсора О*2,
диффундирующего к разл. активным центрам, на к-рых он может либо перейти
в прочно адсорбированный атомарный кислород, либо взаимодействовать, напр.,
с молекулой NH3, адсорбированной на Pt:
Установлено, что число мест на пов-сти для адсорбции О2 в
виде прекурсора значительно больше числа активных центров каталитич. превращения.
Предполагается, что прекурсором м. б. колебательно- или электронно-возбужденная
молекула.
Каталитическое превращение на поверхности. Считается, что катализ
протекает по двум основным механизмам. В случае механизма Ленгмюра-Хиншельвуда
в р-ции участвуют только адсорбиров. частицы, а скорость р-ции пропорциональна
заполнениям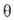 пов-сти (долям пов-сти, занятым адсорбиров. частицами). Для р-ции типа
А + В -> продукты скорость превращения выражается соотношением:
пов-сти (долям пов-сти, занятым адсорбиров. частицами). Для р-ции типа
А + В -> продукты скорость превращения выражается соотношением:

где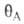 и и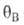 -заполнения
пов-сти молекулами А и В соотв., определяемые в случае однородной пов-сти
и обратимой адсорбции изотермой Ленгмгора (см. Адсорбция). При т.
наз. ударном механизме (механизме Ридила-Или) частица А из газовой фазы
сталкивается с адсорбированной на пов-сти частицей В, образуя продукты
р-ции. В этом случае -заполнения
пов-сти молекулами А и В соотв., определяемые в случае однородной пов-сти
и обратимой адсорбции изотермой Ленгмгора (см. Адсорбция). При т.
наз. ударном механизме (механизме Ридила-Или) частица А из газовой фазы
сталкивается с адсорбированной на пов-сти частицей В, образуя продукты
р-ции. В этом случае
где РА-парциальное давление реагента А.
Однако установлено, что прочно адсорбиров. молекулы могут непосредственно
взаимод. между собой, лишь если они занимают соседние активные центры (соседние
атомы пов-сти катализатора), что в большинстве случаев маловероятно. Как
правило, для Г. к. необходимо, чтобы одна из частиц (напр., А) перешла
в состояние слабой адсорбции и продиффундировала ко второй частице (В).
Элементарной стадией катализа при этом может оказаться именно поверхностная
диффузия. Исследования методом молекулярных пучков показали, что ударный
механизм Г. к. в чистом виде практически не наблюдается. При впуске молекул
А на катализатор с адсорбированными на нем молекулами В (напр., при впуске
Н2 на Pt, покрытую D2) продукт (HD) в отраженном
пучке появляется не сразу, а спустя нек-рое время, необходимое для диффузии
молекул А к активным центрам катализатора. Ур-ние (7) в этом случае соблюдается.
Десорбция. Простая десорбция происходит путем разрыва связи молекулы
продукта с пов-стью. Прочность хим. связи составляет 200-400 кДж/моль и
существенно превышает энергию активации каталитич. р-ции. В условиях Г.
к. часто происходит не простая десорбция, а десорбция через ассоциативный
комплекс, почти не требующая затрат энергии. Напр., гидроксильная группа
на пов-сти А12О3 переходит в газовую фазу при взаимод.
с водяным паром или спиртом по бимолекулярному механизму:

Если группа OR пов-сти разлагается далее на группу ОН и молекулу олефина,
протекает каталитич. дегидратация спирта.
Основные типы гетерогенных катализаторов
В Г. к., как и в др. областях катализа, выделяют два типа р-ций: окислительно-восстановительные,
при к-рых роль катализатора сводится к участию в переносе неспаренных электронов,
и кислотно-основные, при к-рых взаимод. катализатора с реагирующими в-вами
сопровождается переходом протона или электронных пар. Окислит.-восстановит.
Г. к. происходит на пов-сти металлов или полупроводников, т.е. в-в, способных
передавать или принимать неспаренные электроны от реагирующих молекул.
Кислотно-основные р-ции протекают на пов-сти твердых к-т или оснований,
способных передавать или принимать протон от реагентов или же способных
к хим. взаимод. с реагентами без разделения пары электронов. Рассмотрим
возможные механизмы этих взаимодействий.
Катализаторы-полупроводники. Согласно электронной теории Г. к.,
каталитич. активность полупроводников связана с объемной концентрацией
носителей тока (электронов и дырок). Адсорбция частицы на пов-сти полупроводника
приводит к образованию дополнит. (примесного) энергетич. уровня в запрещенной
зоне. Переход электрона или дырки на этот уровень изменяет их объемную
концентрацию и св-ва пов-сти (напр., работу выхода электрона), на к-рой
возникают заряженные центры, участвующие в каталитич. превращении. Можно
представить, напр., что дегидрирование изопропилового спирта происходит
по механизму:
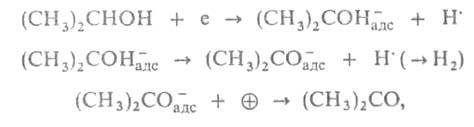
где е-электрон катализатора, -своб.
дырка. Поскольку объемная концентрация носителей тока зависит от положения
уровня Ферми и изменяется при всяком сдвиге последнего, предполагалась
возможность регулирования каталитич. активности полупроводника смещением
уровня Ферми. Дальнейшие исследования, однако, не подтвердили суще" ствования
электронного равновесия между пов-стью и объемом катализатора-полупроводника
в условиях Г. к. Экспериментально установлен ряд корреляций каталитич.
активности полупроводников с проводимостью п- или р-типа
с их св-вами. В частности, известна корреляция константы скорости Г. к.
с шириной запрещенной зоны -своб.
дырка. Поскольку объемная концентрация носителей тока зависит от положения
уровня Ферми и изменяется при всяком сдвиге последнего, предполагалась
возможность регулирования каталитич. активности полупроводника смещением
уровня Ферми. Дальнейшие исследования, однако, не подтвердили суще" ствования
электронного равновесия между пов-стью и объемом катализатора-полупроводника
в условиях Г. к. Экспериментально установлен ряд корреляций каталитич.
активности полупроводников с проводимостью п- или р-типа
с их св-вами. В частности, известна корреляция константы скорости Г. к.
с шириной запрещенной зоны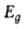 : :

где а и b- эмпирии, постоянные.
Наиб. важную роль в Г. к. играют полупроводники, представляющие собой
соед. элементов VI гр. периодич. системы (О, S, Se, Те) с переходными металлами
(обычно оксиды или сульфиды металлов). Каталитич. св-ва этих в-в определяются
наличием у атомов переходных металлов неск. степеней окисления, к-рые в
условиях катализа легко изменяются в результате переноса электрона от атома
др. реагента. Напр., Мо в оксидах может иметь степени окисления Мо2
+ , Мо3 + , Мо4 +
, Мо5+ и Мо6+, поэтому он легко окисляется и восстанавливается
в условиях Г. к., обеспечивая тем самым каталитич. цикл. Важно также наличие
у поверхностных атомов переходных металлов низкоэнергетич. d-орбиталей
разной симметрии. Это позволяет связать каталитич. активность их оксидов
и сульфидов с электронной конфигурацией J-орбиталей, а также с возможностью
образования промежут. поверхностных соед. типа комплексных. Напр., для
оксидов металлов установлена четкая корреляция между константами скорости
H-D обмена и мн. р-ций окисления с числом d-электронов катиона, т. е. положением
переходного элемента в периодич. системе. Максимальна активность оксидов
с электронной конфигурацией катионов d3 (Cr2O3,
МnО,) и d6-d8 (Co3O4,
NiO), минимальна - с конфигурацией do (TiO2),
d5 (Fe2O3), d10 (ZnO,
Cu2O) (рис. З). Подобная зависимость объясняется кристаллического
поля теорией.
Рис. 3. Изменение каталитич. активности оксидов переходных
металлов в р-ции обмена H2-D2.
Методами ИК-спектроскопии подтверждено, что в ряде р-ций с участием
олефинов (селективное окисление, полимеризация, диспропорционирование)
на пов-сти оксидов переходных металлов с электронной конфигурацией катиона
d1 (Мо5 +, V4 +,
Ti3 +) образуются комплексы
и комплексы
и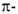 аллильные
комплексы типа I, где М-атом металла. аллильные
комплексы типа I, где М-атом металла.
Катализаторы-металлы. Металлы обычно значительно активнее оксидов и
обладают более универсальным каталитич. действием, хотя, как правило, менее
селективны. наиб. универсальны металлы VIII гр. периодич. системы, особенно
Pt и Pd, катализирующие разл. р-ции окисления, гидрирования, дегидрирования
и т.д. при низких т-рах (комнатной и более низких). Каталитич. активность
определяется электронной конфигурацией и симметрией d-орбиталей
поверхностных атомов. В хим. взаимод. с молекулами реагирующих на пов-сти
в-в участвуют только те d-орбитали, к-рые направлены от пов-сти
наружу и имеют одинаковую группу симметрии с молекулярными орбиталями реагентов.
Участие d-электронов в хим. связи металла с адсорбиров. молекулами подтверждено
методами фотоэлектронной и УФ-спектроскопии для Pt-катализатора.
Металлы, находящиеся в конце переходных периодов, имеют в d-оболочке
дырки (отсутствие электронов), что облегчает их участие в каталитич. превращении.
Металлы, находящиеся в начале периода, обычно образуют прочную связь с
молекулами реагентов. Это приводит к образованию фаз (поверхностных или
объемных) оксидов, гидридов и т. п. и снижению каталитич. активности металла.
Так, Ni активен в р-циях гидрирования, а Си малоактивна. При сплавлении
активных металлов VIII группы с неактивными металлами 1б группы каталитич.
активность уменьшается вследствие заполнения d-оболочки электронами. Напр.,
для сплавов Cu-Ni падение активности наступает при составе 53% Си и 47%
Ni, когда s-электроны Си заполняют J-оболочку Ni.
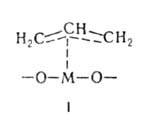
В р-циях с участием Н2 наиб. активны металлы, на пов-сти
к-рых происходит его хемосорбция с диссоциацией и низкой энергией связи
атомарного водорода. Сплавы Cu-Ni, Au-Pt, Ag-Pd менее активны, чем чистые
металлы VIII группы. На чистых металлах 1б группы Н2 не адсорбируется
и не активируется.
В пром-сти широко применяют мелкодисперсные метал-лич. катализаторы,
нанесенные на носители (SiO2, A12O3, алюмосиликаты,
активный уголь, кизельгур и др.). Это повышает пов-сть катализатора, уменьшает
его расход, предохраняет частицы от спекания. По мере уменьшения размера
частиц (повышения дисперсности) катализатора его активность в одних р-циях
остается неизменной (структурно-нечувствительные или "незатрудненные" р-ции),
в других р-циях на частицах размером 2-4 нм при общем росте активности
наблюдается снижение числа оборотов р-ции; это т. наз. структурно-чувствительные
или "затрудненные" р-ции. Так, большинство р-ций гидрирования олефинов
и ароматич. соед. являются структурно-нечувствительными; р-ции синтеза
NH3, гидрогенолиза связи С—С, окисления, изомеризации и др.
структурно-чувствительны. Различие между этими типами гетерогенно-каталитич.
р-ций на металлах объясняют тем, что в первом из них активным центром является
каждый атом пов-сти, во втором-совокупность неск. атомов. Разбавление в
катализаторе-сплаве активного металла неактивным компонентом не влияет
на его активность в структурно-нечувствит. р-циях и приводит к значит,
ее снижению в структурно-чувствит. р-циях. Кроме того, в структурно-чувствительных
р-циях активность катализатора зависит от выхода на пов-сть кристаллич.
граней, ребер, концентрации дислокаций, Так, в синтезе NH3 на
монокристаллах Fe наиб. активностью обладает грань (111), в гидрогенолизе
углеводородов на Pt и Pd-ребра монокристаллов; необходимый для р-ции Н2
диссоциирует на ребрах и диффундирует к активным центрам, на к-рых происходит
каталитич. превращение. Каталитич. активность металлов на носителях изменяется
также вследствие их хим. взаимод. с носителем (см. Нанесенные катализаторы).
Катализ на твердых кислотах и основаниях. Для катализаторов кислотно-основного
типа специфика твердого тела не выражена так резко, как для полупроводников
и металлов. Активные центры кислотных кат. представляют подвижные протоны
Н (центры Бренстеда) или атомы, способные присоединять пару электронов
(центры Льюиса), напр. атом А1 на пов-сти А12О3.
Соотв. основными центрами являются акцепторы протона или доноры электронной
пары, напр. атомы кислорода на пов-сти CaO, MgO и т.п. Кислотными бренстедовскими
центрами простых оксидов металлов являются поверхностные гидроксильные
группы, остающиеся после частичной дегидратации пов-сти при нагр., или
молекулы Н2О, координационно связанные с пов-стью. Для металла
М, находящегося в начале каждого периода, гидроксильные группы имеют основные
св-ва [...ОМ]+ [ОН]-, для находящегося в конце периода-кислотные:
[...ОМО]-Н + . Льюисовскими кислотными центрами служат
координационно-ненасыщенные ионы, напр. АlO-2 на
А12О3. Эти центры способны взаимод. с реагирующей
молекулой-донором пары электронов. Кислотными катализаторами являются оксиды
металлов с большим отношением заряда иона к его радиусу-оксиды Мо, Zn,
Ca, Pb и др. Их активность связана с положением металла в периодич. системе
и возрастает в периодах при переходе к V-VII группам, а в группах-при переходе
к I периоду.
Смешанные катализаторы. В р-циях кислотно-основного типа (крекинг, дегидратация,
изомеризация и др.) высокой активностью обладают катализаторы, состоящие
из неск. в-в,- оксиды металлов с разл. зарядом катиона, аморфные алюмосиликаты
и цеолиты, гетерополикислоты, сульфаты, фосфаты и др. Именно на пов-сти
смешанных систем легче образуются реакционноспособные заряженные частицы.
Напр., в алюмосиликатах ион А13+ замещает Si4+ в
кремнекислородной решетке; меньший заряд Al3+ по сравнению с
Si4+ компенсируется появлением центра Бренстеда Н+.
Присоединение образовавшегося Н + к реагентам приводит к возникновению
заряженных реакционноспособных частиц, напр. карбкатионов (СН3СН=СН2
+ Н+ -> C3H7+), участвующих
далее в катализе. На основных центрах образуются отрицательно заряженные
частицы, в р-циях углеводородов-карбанионы.
Полифункциональные катализаторы. Пром. каталитич. процессы часто проводят
на катализаторах, сочетающих разл. ф-ции. Напр., превращения углеводородов
в риформинге ускоряются катализаторами, в к-рых переходные металлы, гл.обр.
Pt или Ni, комбинируются с кислотным оксидом, напр. алюмосиликатом или
А12О3, модифицированным фтором. В этом случае Pt
оказывает дегидрирующее действие, а кислотный оксид-изомеризующее. Катализ
протекает вблизи границы раздела фаз или в результате перемещения активной
частицы из одной фазы в другую:
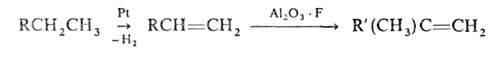
Полифункциональные кат., как правило, состоят из неск. фаз, каждая из
к-рых ускоряет одну из стадий сложного процесса. Более 50 лет назад С.
В. Лебедев совм. с сотрудниками подобрал селективный кат. синтеза бутадиена
из спирта по р-ции:
2С2Н5ОН -> СН2=СНСН=СН2
+ 2Н2О + Н2
Были выделены стадии процесса: дегидратация, дегидрирование и конденсация
и для каждой из стадий подобран свой катализатор (активная глина, ZnO и
MgO соотв.). В смешанном кат. эти компоненты находятся в соотношении, обеспечивающем
макс. выход бутадиена.
Высокоселективные кат. парциального окисления представляют смесь оксидов
разл. металлов. Напр., для окисления пропилена в акролеин применяют катализатор,
состоящий из оксидов Bi, Mo, Fe, Co и др. В этом катализаторе Bi2(MoO4)
служит для адсорбции и активации пропилена, FеМоО4-для активации
О2, на дефектах кристаллич. структуры Fe2(MoO4)3
происходит перенос ионов кислорода от центров его адсорбции к центрам адсорбции
олефина, СоМоО4 служит для стабилизации структуры FeMoO4.
Гетерогенизированньзе металлокомплексные катализаторы. В 70-80-е гг.
20 в. широко исследуются катализаторы-комплексы металлов, закрепленные
на пов-сти носителя (SiO2, А12О3 и др.).
Состав таких комплексов описывается общей ф-лой XnMmYy,
где М-активный центр (атом) переходного металла, Х-лиганд, связывающий
атом металла с пов-стью, Y-внеш. лиганд. В общем случае комплекс м. б.
моноядерным (т = 1) или полиядерным (т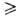 2)
и связан с пов-стью одним или неск. лигандами X. Напр., растворимый комплексный
катализатор гидрирования Rh[P(C6H5)3]3Cl
м. б. закреплен на пов-сти силикагеля: 2)
и связан с пов-стью одним или неск. лигандами X. Напр., растворимый комплексный
катализатор гидрирования Rh[P(C6H5)3]3Cl
м. б. закреплен на пов-сти силикагеля:
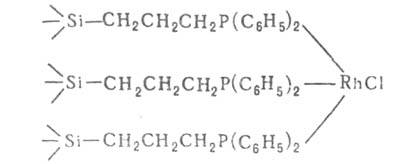
При взаимод. металлоорг. соед. с ОН-группами носителя можно получить
закрепленные комплексы, не имеющие р-римых аналогов. Гетерогенизированные
металлокомплексные кат. сочетают высокую активность и однородность по каталитич.
св-вам, характерные для гомог. катализа, с удобствами технол. применения
гетерог. катализаторов. Получены Гетерогенизированные кат., содержащие
комплексы Ti, Zr, Mo, W, Ni и др. металлов, активные в полимеризации, окислении,
гидрировании и др. Технол. применение гетерогенизированных комплексных
кат. пока невелико из-за нестабильности гетерогенизиров. комплексов в условиях
катализа и трудности их регенерации. Гетерогенизиров. комплексы используют
для получения нанесенных металлич. катализаторов очень высокой дисперсности
(размер частички - кластера - неск. атомов).
Технологические особенности процессов
В пром-сти наиб. распространены реакторы с неподвижным слоем катализатора,
в к-рых через слой гранулиров. или таблетированного кат. пропускается (обычно
сверху вниз) поток газовых, иногда жидких, реагентов. Катализаторы, используемые
в этих реакторах, кроме необходимой активности и селективности, должны
обладать достаточной прочностью к истиранию, т.к. истирание увеличивает
гидравлич. сопротивление слоя. Высокая уд. пов-сть и пористость катализатора
повышают его общую активность, однако способствуют диффузионному торможению
р-ции. Диффузионные процессы особенно вредны в случае последоват. каталитич.
р-ций, когда в результате диффузионных затруднений с отводом продукта последний
может претерпевать нежелательные дальнейшие превращения. Напр., при окислении
этилена в этиленоксид на пористом кат. селективность может ухудшаться в
результате доокис-ления С2Н4О. Для ликвидации диффузионных
осложнений применяют непористые носители или дробят зерна катализатора.
Жидкофазный Г.к. проводят в реакторах смешения, в к-рых мелко зернистый
кат. суспендируют в среде реагентов или р-рителя. Такие реакторы используют
как в периодич., так и в непрерывном режимах. Для устранения внешне-диффузионных
осложнений смесь обычно интенсивно перемешивают. По окончании р-ции катализатор
необходимо отделить от реагентов.
Применяют также реакторы с кипящим, или псевдоожиженным, слоем катализатора,
в к-рых пылевидный катализатор поднимается восходящим потоком жидкости
или газа. Преимущества Г. к. в псевдоожиженном слое - возможность использования
мелкодисперсных непористых частиц, что снижает влияние внутр. диффузии,
непрерывное удаление отработанного катализатора и возможность его замены,
высокий коэф. теплопередачи, позволяющий поддерживать постоянную т-ру по
всему объему кипящего слоя. Псевдоожиженный слой используют для р-ций с
интенсивным тепловыделением, напр. при каталитич. окислении. К его недостаткам
относятся повышенная истираемость катализатора и вынос частиц катализатора
из реактора, к-рые затем необходимо улавливать.
В нек-рых процессах, напр. риформинге, применяют реакторы с движущимся
слоем гранулиров. катализатора, в к-рый постоянно подается свежий катализатор,
а отработанный катализатор идет на регенерацию.
Для конструирования реакторов Г. к. необходимо разработать кинетич.
модель процесса, к-рая позволяет определить требуемое кол-во катализатора
и объем реактора, обеспечивающий макс. скорость р-ции и выход продукта.
Расчеты реакторов должны учитывать также явления тепло- и массопереноса.
При осуществлении экзотермич. р-ций часто используют проточно-циркуляц.
схемы, включающие теплообменники между слоями катализатора. Расчеты пром.
реакторов основываются на методах макрокинетики.
Исторический очерк
Научные исследования Г.к. начались в кон. 19-нач. 20 вв. с работ по
дегидратации спиртов на глинах и по разложению аммиака и пероксида водорода
на разл. твердых телах. В 19 в. открыты мн. гетерог.-каталитич. процессы,
с 20 в. началось активное изучение механизма Г. к. Это определялось потребностями
развития хим. технологии, прежде всего процессов получения минер. к-т и
аммиака, а затем и потребностями нефтепереработки и нефтесинтеза. Пониманию
природы Г. к. способствовало применение физ. и кинетич. методов к исследованию
гетерог.-каталитич. систем.
Наиб. плодотворной для создания совр. представлений о Г. к. явилась
идея Д. Менделеева (позднее развитая Н. Д. Зелинским) о совместном воздействии
физ. и хим. св-в пов-сти катализатора на превращаемые молекулы. Еще в сер.
19 в. А. И. Ходневым было выдвинуто представление об образовании промежут.
поверхностных соед., роль к-рых наиб. последовательно рассмотрена в кон.
19-нач. 20 вв. П. Сабатье. Важную роль в развитии теоретич. представлений
сыграло выдвинутое Г. Тейлором в 1925-26 предположение, связывающее каталитич.
активность твердых тел с расположением атомов на их пов-сти и наличием
активных центров. Мультиплетная теория Г. к. (А. А. Баландин, первые публикации
1929) придает решающее значение соответствию расстояний между атомами молекул
реагентов и параметров кристаллич. структуры катализатора (металла). В
дальнейшем теория дополнена представлением о необходимости определенного
соответствия энергий связей, разрывающихся и образующихся в результате
р-ции, и энергий связи реагентов с катализатором при промежут. взаимодействии.
Каталитич. действие полупроводников объясняли на основе электронной теории,
согласно к-рой взаимод. реагентов с катализатором осуществляется при участии
электронов проводимости и потому зависит от расположения энергетич. зон
и локальных уровней, концентрации носителей тока, работы выхода электрона
и т.п. Широкое распространение получило предположение, согласно к-рому
особыми активными местами на пов-сти твердых катализаторов являются кристаллографич.
ребра и углы, а также выходы на пов-сть дислокаций, т.е. нарушения кристаллич.
структуры. Для нанесенных катализаторов были развиты представления об особых
св-вах отдельно расположенных, локализованных на пов-сти атомов или совокупностей
атомов-ансамблей (теория активных ансамблей Н.И. Кобозева, 1939).
С 60-х гг. промежут. хим. взаимод. рассматривается преим. как локальное,
определяемое электронной структурой отд. атомов или ионов активного компонента
катализатора с учетом влияния окружения. При этом используются квантовохим.
теории кристаллич. поля и поля лигандов, успешно применяемые в химии комплексных
соед. Совр. теории Г. к. основаны на квантовохим. расчетах структуры и
реакционной способности образующихся на пов-сти комплексов и эксперим.
исследовании кинетики элементарных стадий.
===
Исп. литература для статьи «ГЕТЕРОГЕННЫЙ КАТАЛИЗ»: Томас Дж., Томас У., Гетерогенный катализ, пер. с англ.,
М., 1969; Киперман С Л., Основы химической кинетики в гетерогенном катализе,
М., 1979; Крылов О. В., "Кинетика и катализ", 1980, т. 21, № 1, с. 79-95;
Крылов О. В., Киселев В. Ф., Адсорбция и катализ на переходных металлах
и их оксидах, М., 1981; Catalysis. Science and technology, ed. by J. R.
Anderson, M. Boudart, v. 1-5, В., 1981-84. О.В.Крылов.
Страница «ГЕТЕРОГЕННЫЙ КАТАЛИЗ» подготовлена по материалам химической энциклопедии.
|



