КАЧЕСТВЕННЫЙ АНАЛИЗ, идентификация (обнаружение) компонентов анализируемых в-в и приблизительная количеств, оценка их содержания в в-вах и материалах. В качестве компонентов м. б. атомы и ионы, изотопы элементов и отдельные нуклиды, молекулы, функц. группы и радикалы, фазы (см. Элементный анализ, Изотопный анализ. Молекулярный анализ. Органических веществ анализ, Фазовый анализ).
Первоначально К. а. возник как вид органолептич. восприятия продуктов потребления и произ-ва для оценки их качества. В первую очередь это относилось к лек. в-вам, для анализа к-рых был разработан т. наз. мокрый путь, т. е. анализ жидкостей и р-ров. С переходом к произ-ву и применению металлов возник пробирный анализ, первоначально как К. а. для определения подлинности благородных металлов. В дальнейшем он стал методом приближенного количеств, анализа. Одновременно развивались разл. варианты пирохим. К. а. для определения цветных металлов, железа, а также для анализа содержащих металлы минералов и руд. Качеств. аналит. сигналом при этом служили внеш. вид королька восстановленного металла, окраска конденсатов выделяющихся летучих продуктов, образование характерно окрашенных стекол ("перлов") при сплавлении анализируемых в-в с содой, бурой или селитрой.
В основу анализа орг. соед. еще А. Лавуазье положил процессы сожжения с образованием СО2 и Н2О. Далее этот метод был развит др. учеными на основе тех же пирохим. процессов и газового анализа, причем К. а. тесно слился с количественным. После открытия изомерии в К. а. было включено изучение хим. структуры орг. соединений. Классический орг. анализ - родоначальник микрохим. методов анализа и автоматич. анализаторов.
Параллельно с хим. методами К. а. развивались и чисто физические - от метода установления хим. состава бинарного сплава путем измерения уд. веса (метод Архимеда) до спектроскопии, измерения эдс, поверхностного натяжения р-ров и т. д. С сер. 20 в. значение физ. методов К. а. неизмеримо возросло.
Качеств. и количеств, анализы развивались в тесном взаимод., т. к. только при уточнении количеств. данных возможна полная расшифровка качеств. компонентного состава в-ва, а на основе данных К. а. - совершенствование количеств. анализа. При этом К. а. строится на основе возрастающей дифференциации св-в компонентов, а количественный - на возможности воспринимать и дифференцировать аналит. сигналы миним. интенсивности.
Хим. методы элементного анализа неорг. соединений. Основаны на ионных р-циях и позволяют обнаруживать элементы в форме катионов и анионов. Для К. а. катионов используют разл. схемы систематич. анализа с последоват. разделением катионов на группы и подгруппы, внутри к-рых возможна идентификация отдельных элементов. Аналит. группы обычно именуют по групповому реагенту.
1. Группа соляной к-ты; в нее входят Ag, Hg(I), Pb, Tl(I), к-рые образуют хлориды, малорастворимые в воде и кислых р-рах, а также W, Nb, Та, образующие в этих же условиях малорастворимые гидраты оксидов.
2. Группа гидразина; в нее входят Pd, Pt, Au, Se, Те, к-рые восстанавливаются в кислом р-ре; при отсутствии благородных металлов эта группа опускается, a Se и Те переходят в следующую.
3. Группа сероводорода. Подразделяют на три подгруппы: а) меди - Сu, Pb, Hg(II), Bi, Cd; образуют сульфиды, нерастворимые в полисульфиде аммония (NH4)2Sn; б) мышьяка - As, Sb, Sn; образуют тиосоли, р-римые в (NH4)2Sn; в) более редких элементов - Ge, Se, Те, Mo; образуют сульфиды, р-римые в (МН4)2Sn:
4. Группа (NH4)2S - уротропина, элементы к-рой образуют сульфиды или гидроксиды, малорастворимые в
аммиачном р-ре (NH4)2S. Подразделяют условно на три подгруппы: а) элементы со степенью окисления +2-Со, Ni, Mn, Zn; б) элементы со степенью окисления + 3 - Fe, A1, Сr; в) др. элементы - Be, Ga, In, Sc, Y, La, Th, U, Ti, Zr, Hf, Nb, Та (при наличии катионов этой подгруппы предварительно отделяют элементы с высокими степенями окисления обработкой уротропином в слабокислой среде). К этой группе относят также V, W, образующие первоначально р-римые тиосоли, разрушающиеся при подкислении.
5. Группа (NH4)2CO3; в нее входят Са, Sr, Ba, к-рые образуют карбонаты, малорастворимые в аммиачной среде, и не образуют осадков с описанными групповыми реагентами.
6. Группа р-римых соед., не образующих осадков со всеми указанными групповыми реагентами, - Li, Na, К, Mg, Rb, Cs. В учебных курсах нумерацию групп часто обращают, начиная ее с группы р-римых соединений.
Недостатки описанной схемы: плохое отделение Zn2+ от Cd2+, неточное разделение Sn2+, Рb2+, соосаждение нек-рых сульфидов четвертой группы (Fe и Zn) с CuS, окисление сульфидов в р-римые сульфаты и др., а также высокая токсичность H2S. Имеются бессероводородные методы систематич. К. а. К ним относятся методы с применением заменителей H2S, дающих ион S2- в водных р-рах (тиомочевина, тиоацетамид, тиосульфат), и наиб, распространенные методы без иона S2-: кислотно-щелочной, аммиачно-фосфатный, гидразин-гидроксиламиновый, фторидно-бензоатный и др. Напр., в кислотно-щелочном методе катионы разделяют на группы малорастворимых хлоридов или сульфатов, амфотерных гидроксидов, нерастворимых в щелочах гидроксидов, амминокомплексов, растворимых в воде солей.
Полные схемы систематич. К. а. осуществляются редко. Обычно их используют частично в соответствии с конкретным набором ионов, для разделения ионов металлов в количеств. анализе, а также в учебных курсах аналит. химии.
К. а. смесей неметаллов (исключая анализ орг. в-в) осуществляют путем идентификации анионов в водных и водно-орг. средах. Анионы не имеют общеустановленного разделения на группы, число к-рых значительно варьирует в разных схемах анализа. Обычно анионы классифицируют по признаку растворимости солей (табл. 1) и по признаку окислит.-восстановит. активности (табл. 2). Групповые

реагенты в анализе анионов служат только для их обнаружения (в отличие от катионов, где такие реактивы служат и для разделения). Для отделения катионов, мешающих обнаружению анионов, анализируемый р-р предварительно обрабатывают 1 М р-ром соды для осаждения карбонатов, гидроксокарбонатов и гидроксидов тяжелых металлов (на карбонат-ион, возможно имеющийся в пробе, проводят предварит. испытание).
При наличии в анализируемом в-ве ионов, для к-рых существуют селективные реагенты, их обнаружение проводят из исходного р-ра с помощью характерных индивидуальных

р-ций (дробный метод). При этом обычно сначала изолируют мешающие компоненты осаждением или маскированием, а затем специфич. р-цией идентифицируют искомый ион. Основой для создания дробного анализа послужило получение большого набора реагентов органических на ионы неорг. в-в, а также разработка техники капельного анализа. Разработан дробный метод полного К. а. катионов и анионов.
Техника проведения К. а. развивается в направлении отказа от макрометодов и перехода к полумикро- (100-10 мг), микро- (10-0,1 мг) и ультрамикрометодам (менее 0,1 мг). Полумикроанализ широко применяют в учебной работе; микро- и ультрамикроанализ - при исследовании биол. объектов, а также в электронной технике, особенно полупроводниковой, и радиохимии.
Количеств, характеристика методик К. а. - предел обнаружения, т.е. миним. кол-во искомого компонента (в мкг или нг), к-рое м. б. надежно идентифицировано: для р-ров используется величина предельной концентрации Сx, min или обратная ей величина предельного разбавления Dх (предельный объем р-ра, к-рый приходится на 1 мкг определяемого компонента). Предел обнаружения и Сх, min связаны друг с другом выражением:
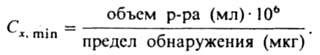
Иногда пользуются величиной pDx=-lgDx; для большинства микрохим. р-ций К. a. pDx = 4-6.
Хим. методы К. а. орг. соединений. В составе орг. сосд. обычно идентифицируют С, Н, О, N, S, Р, галогены и др. Углерод - по СО2, образующемуся после сжигания пробы в раскаленной трубке в присут. СuО; водород - по Н2О, конденсирующейся на холодных участках трубки, или по H2S, к-рый образуется после прокаливания пробы с безводными Na2SO3 и Na,S2O3 и обнаруживается бумагой, пропитанной р-ром (СН3СОО)2Рb или нитропруссида натрия. Азот, серу и галогены определяют после разложения в-ва расплавленным К или Na в открытых или запаянных стеклянных трубках по качеств, р-циям в р-ре на образующиеся KCN (или NaCN), сульфиды, тиоцианаты, цианаты и галогениды; одновременно определяют наличие углерода по остатку на фильтре. Кислород обычно идентифицируют по функц. группам (карбонильной, альдегидной и др.); для прямого обнаружения пробу нагревают в токе N2 или Н2 в присут. платинового катализатора и идентифицируют по СО2 и Н2О. Фосфор обнаруживают по р-ции с (МН4)2МоО4, а мышьяк-по р-ции с H2S после оглавления исследуемого в-ва с содой или селитрой и обработки остатка НСl. Металлы идентифицируют в зольном остатке после сожжения пробы.
Ряд специфич. и чувствительных р-ций для элементного анализа орг. соед. предложен в системе капельного анализа с помощью цветных р-ций.
Приборами для одновременного качеств, и количеств, элементного анализа служат автоматич. анализаторы типов CHN, CHNS, ClBrICHNS, к-рые снабжены специфич. сорбционными или серийными хроматографич. устройствами для разделения продуктов разложения и детекторами для их идентификации.
Важный метод исследования орг. соед.-функциональный К. а., т. е. обнаружение атомов или групп атомов, определяющих строение данного класса орг. соед. и их конкретные св-ва.
Хим. методы К. а. имеют практич. значение при необходимости обнаружения только неск. элементов. Для многоэлементного К. а. применяют физ.-хим. методы, такие как хроматография, электрохим. методы, в осн. полярография, и др. и физические методы, напр, атомно-эмиссионную спектрометрию (см. Спектральный анализ)(предел обнаружения 1 мкг на 1 г твердой пробы или 1 мл р-ра), атомно-абсорбционный анализ (предел обнаружения порядка пикограммов), рентгеноэмиссионный и рентгенофлуоресцентный анализ (см. Рентгеновская спектроскопия)(миним. анализируемый объем 1 мкм3, предел обнаружения 10-2-10-3% по массе).
Молекулярный и функциональный К. а. проводят с помощью инфракрасной спектроскопии, комбинационного рассеяния спектроскопии, ядерного магнитного резонанса, электронного парамагнитного резонанса. Особое место в совр. К. а. занимает масс-спектрометрия и хромато-масс-спектрометрия (ниж. предел обнаружения - 10-7% по массе).
В основе фазового К. а. лежат процессы выделения отдельных фаз из сплава или руды и установление их состава хим. или физ.-хим. методами. Наиб. значение имеет рентгеновский фазовый анализ и термогравиметрия (особенно при анализе минералов). К. а. и полуколич. анализ фаз в гетерофазной системе возможно также осуществить на шлифе образца посредством электронного микрозонда.
Для анализа нуклидов используют активационный анализ.
В совр. неорг. К. а. ведущая роль принадлежит физ. методам, к-рые позволяют решать задачи идентификации и установления строения хим. соед., определения их локализации в объекте, установления типа хим. связи между атомами и группами атомов; в орг. К. а. хим. и физ. методы используются комплексно.
===
Исп. литература для статьи «КАЧЕСТВЕННЫЙ АНАЛИЗ»: Губен-Вейль, Методы органической химии, т. 2 - Методы анализа, 2 изд., М., 1967; Мурашова В. И., Тананаева А.Н., Ховякова Р.Ф., Качественный химический дробный анализ, М., 1976; Ляликов Ю.С., Клячко Ю.А., Теоретические основы современного качественного анализа, М., 1978; Гельман Н. Э.. Кипаренко Л. М., Автоматический элементный анализ органических соединений, "Ж. Всес. хим. об-ва им. Д.И. Менделеева", 1980, т. 25, №6, с. 641-51; Идентификация органических соединений, пер. с англ., М., 1983, с. 100-15: Иоффе Б. В., Костиков Р. Р., Разин В. В., Физические методы определения строения органических соединений, М., 1984; М азор Л.. Методы органического анализа, пер. с англ., М., 1986. Ю.А.Клячко.
Страница «КАЧЕСТВЕННЫЙ АНАЛИЗ» подготовлена по материалам химической энциклопедии.
|



