КОЛЕБАТЕЛЬНЫЕ СПЕКТРЫ, мол. спектры, обусловленные квантовыми переходами между колебат. уровнями энергии молекул. Экспериментально наблюдаются как ИК спектры поглощения и спектры комбинац. рассеяния (КР); диапазон волновых чисел ~10-4000 см-1 (частоты колебат. переходов 3.1011-1014 Гц). Колебат. уровни энергии определяются квантованием колебат. движения атомных ядер.
Двухатомные молекулы. В простейшем случае двухатомную молекулу представляют моделью двух взаимодействующих точечных масс m1 и m2 с равновесным расстоянием rе между ними (длина связи), а колебат. движение ядер считается гармоническим и описывается единств, координатой q=r-re, где r - текущее межъядерное расстояние. Зависимость потенциальной энергии колебат. движения V от q определяют в приближении гармонич. осциллятора [колеблющаяся материальная точка с приведенной массой m=m1m2/(m1+m2)] как ф-цию V=l/2(Keq2), где Ке=(d2V/dq2)q=0 - гармонич. силовая постоянная
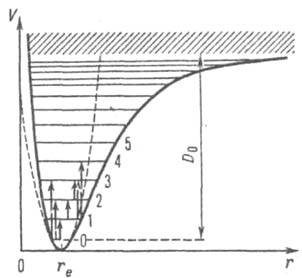
Рис. 1. Зависимость потенциальной энергии V гармонич. осциллятора (пунктирная кривая) и реальной двухатомной молекулы (сплошная кривая) от межъядерного расстояния r (rс равновесное значение r); горизонтальными прямыми линиями показаны колебат. уровни (0, 1, 2, ... значения колебат. квантового числа), вертикальными стрелками - нек-рые колебат. переходы; D0 - энергия диссоциации молекулы; заштрихованная область отвечает сплошному спектру. молекулы (пунктирная кривая на рис. 1).
Согласно классич. механике, частота гармонич. колебаний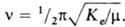 Квантовомех. рассмотрение такой системы дает дискретную последовательность равноотстоящих уровней энергии E(v)=hve(v+1/2), где v = 0, 1, 2, 3, ... - колебательное квантовое число, ve - гармонич. колебательная постоянная молекулы (h - постоянная Планка). При переходе между соседними уровнями, согласно правилу отбора Dv=1, поглощается фотон с энергией hv=DE=E(v+1)-E(v)=hve(v+1+1/2)-hve(v+1/2)=hve, т. е. частота перехода между двумя любыми соседними уровнями всегда одна и та же, причем совпадает с классич. частотой гармонич. колебаний. Поэтому ve наз. также гармонич. частотой.
Для реальных молекул кривая потенциальной энергии не является указанной квадратичной ф-циeй q, т. е. параболой. Колебат. уровни все более сближаются по мере приближения к пределу диссоциации молекулы и для модели ангармонич. осциллятора описываются ур-нием: E(v)= Квантовомех. рассмотрение такой системы дает дискретную последовательность равноотстоящих уровней энергии E(v)=hve(v+1/2), где v = 0, 1, 2, 3, ... - колебательное квантовое число, ve - гармонич. колебательная постоянная молекулы (h - постоянная Планка). При переходе между соседними уровнями, согласно правилу отбора Dv=1, поглощается фотон с энергией hv=DE=E(v+1)-E(v)=hve(v+1+1/2)-hve(v+1/2)=hve, т. е. частота перехода между двумя любыми соседними уровнями всегда одна и та же, причем совпадает с классич. частотой гармонич. колебаний. Поэтому ve наз. также гармонич. частотой.
Для реальных молекул кривая потенциальной энергии не является указанной квадратичной ф-циeй q, т. е. параболой. Колебат. уровни все более сближаются по мере приближения к пределу диссоциации молекулы и для модели ангармонич. осциллятора описываются ур-нием: E(v)=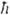 [ve(v+1/2)-X1(v+1/2)2], где X1 - первая постоянная
ангармоничности. Частота перехода между соседними уровнями не остается постоянной, и, кроме того, возможны переходы, отвечающие правилам отбора Dv=2, 3, .... Частота перехода с уровня v=0 на уровень v=1 наз. основной, или фундаментальной, частотой, переходы с уровня v=0 на уровни v>1 дают обертонные частоты, а переходы с уровней v>0 - т. наз. горячие частоты.
В ИК спектре поглощения двухатомных молекул колебат. частоты наблюдаются только у гетероядерных молекул (НСl, NO, CO и т.п.), причем правила отбора определяются изменением их электрич. дипольного момента при колебаниях. В спектрах КР колебат. частоты наблюдаются для любых двухатомных молекул, как гомоядерных, так и гетероядерных (N2, O2, CN и т.п.), т.к. для таких спектров правила отбора определяются изменением поляризуемости молекул при колебаниях. Определяемые из К. с. гармонич. постоянные Ке и ve, постоянные ангармоничности, а также энергия диссоциации D0 - важные характеристики молекулы, необходимые, в частности, для термохим. расчетов. Изучение колебательно-вращат. спектров газов и паров позволяет определять вращат. постоянные Вv (см. Вращательные спектры), моменты инерции и межъядерные расстояния двухатомных молекул.
Многоатомные молекулы рассматривают как системы связанных точечных масс. Колебат. движение ядер относительно равновесных положений при неподвижном центре масс в отсутствие вращения молекулы как целого описывают обычно с использованием т. наз. внутр. естеств. координат qi, выбираемых как изменения длин связей, валентных и двугранных углов пространств, модели молекулы. У молекулы, состоящей из N атомов, имеется n=3N — 6 (у линейной молекулы 3N — 5) колебат. степеней свободы. В пространстве естеств. координат qi сложное колебат. движение ядер можно представить п отдельными колебаниями, каждое с определенной частотой vk (k принимает значения от 1 до n), с к-рой меняются все естеств. координаты qi при определенных для данного колебания амплитудах q0i и фазах. Такие колебания наз. нормальными. Напр., трехатомная линейная молекула АХ2 имеет три нормальных колебания: [ve(v+1/2)-X1(v+1/2)2], где X1 - первая постоянная
ангармоничности. Частота перехода между соседними уровнями не остается постоянной, и, кроме того, возможны переходы, отвечающие правилам отбора Dv=2, 3, .... Частота перехода с уровня v=0 на уровень v=1 наз. основной, или фундаментальной, частотой, переходы с уровня v=0 на уровни v>1 дают обертонные частоты, а переходы с уровней v>0 - т. наз. горячие частоты.
В ИК спектре поглощения двухатомных молекул колебат. частоты наблюдаются только у гетероядерных молекул (НСl, NO, CO и т.п.), причем правила отбора определяются изменением их электрич. дипольного момента при колебаниях. В спектрах КР колебат. частоты наблюдаются для любых двухатомных молекул, как гомоядерных, так и гетероядерных (N2, O2, CN и т.п.), т.к. для таких спектров правила отбора определяются изменением поляризуемости молекул при колебаниях. Определяемые из К. с. гармонич. постоянные Ке и ve, постоянные ангармоничности, а также энергия диссоциации D0 - важные характеристики молекулы, необходимые, в частности, для термохим. расчетов. Изучение колебательно-вращат. спектров газов и паров позволяет определять вращат. постоянные Вv (см. Вращательные спектры), моменты инерции и межъядерные расстояния двухатомных молекул.
Многоатомные молекулы рассматривают как системы связанных точечных масс. Колебат. движение ядер относительно равновесных положений при неподвижном центре масс в отсутствие вращения молекулы как целого описывают обычно с использованием т. наз. внутр. естеств. координат qi, выбираемых как изменения длин связей, валентных и двугранных углов пространств, модели молекулы. У молекулы, состоящей из N атомов, имеется n=3N — 6 (у линейной молекулы 3N — 5) колебат. степеней свободы. В пространстве естеств. координат qi сложное колебат. движение ядер можно представить п отдельными колебаниями, каждое с определенной частотой vk (k принимает значения от 1 до n), с к-рой меняются все естеств. координаты qi при определенных для данного колебания амплитудах q0i и фазах. Такие колебания наз. нормальными. Напр., трехатомная линейная молекула АХ2 имеет три нормальных колебания:
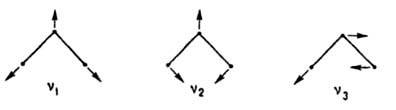
Колебание v1 наз. симметричным валентным колебанием (растяжения связей), v2 - дeфopмaциoнным колебанием (изменение валентного угла), v3 антисимметричным валентным колебанием. В более сложных молекулах встречаются и др. нормальные колебания (изменения двугранных углов, крутильные колебания, пульсации циклов и т.п.).
Квантование колебат. энергии многоатомной молекулы в приближении многомерного гармонич. осциллятора приводит к след, системе колебат. уровней энергии:
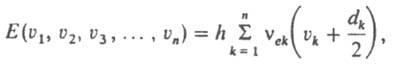
где vek - гармонич. колебат. постоянные, vk - колебат. квантовые числа, dk - степень вырождения уровня энергии по k-му колебат. квантовому числу. Осн. частоты в К. с. обусловлены переходами с нулевого уровня [все vk=0, колебат. энергия на уровни, характеризуемые
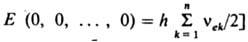
такими наборами квантовых чисел vk, в к-рых только одно из них равно 1, а все остальные равны 0. Как и в случае двухатомных молекул, в ангармонич. приближении возможны также обертонные и "горячие" переходы и, кроме того, т. наз. комбинированные, или
составные, переходы с участием уровней, для к-рых отличны от нуля два или более из квантовых чисел vk (рис. 2).
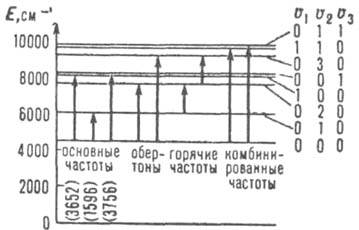
Рис. 2. Система колебат. термов E/hc (см'; с - скорость света) молекулы Н2О и нeк-рые переходы; v1, v2. v3 - колебат. квантовые числа.
Интерпретация и применение. К. с. многоатомных молекул отличаются высокой специфичностью и представляют сложную картину, хотя общее число экспериментально наблюдаемых полос м. б. существенно меньше возможного их числа, теоретически отвечающего предсказываемому набору уровней. Обычно осн. частотам соответствуют более интенсивные полосы в К. с. Правила отбора и вероятность переходов в ИК и КР спектрах различны, т.к. связаны соотв. с изменениями электрич. дипольного момента и поляризуемости молекулы при каждом нормальном колебании. Поэтому появление и интенсивность полос в ИК и КР спектрах по-разному зависит от типа симметрии колебаний (отношения конфигураций молекулы, возникающих в результате колебаний ядер, к операциям симметрии, характеризующим ее равновесную конфигурацию). Нек-рые из полос К. с. могут наблюдаться только в ИК или только в КР спектре, другие - с разной интенсивностью в обоих спектрах, а нек-рые вообще экспериментально не наблюдаются. Так, для молекул, не обладающих симметрией или имеющих низкую симметрию без центра инверсии, все осн. частоты наблюдаются с разной интенсивностью в обоих спектрах, у молекул с центром инверсии ни одна из наблюдаемых частот не повторяется в ИК и КР спектрах (правило альтернативного запрета); нек-рые из частот могут отсутствовать в обоих спектрах. Поэтому важнейшее из применений К. с. - определение симметрии молекулы из сопоставления ИК и КР спектров, наряду с использованием др. эксперим. данных. Задаваясь моделями молекулы с разной симметрией, можно заранее теоретически рассчитать для каждой из моделей, сколько частот в ИК и КР спектрах должно наблюдаться, и на основании сопоставления с эксперим. данными сделать соответствующий выбор модели.
Хотя каждое нормальное колебание, по определению, является колебат. движением всей молекулы, нек-рые из них, особенно у больших молекул, могут более всего затрагивать лишь к.-л. фрагмент молекулы. Амплитуды смещения ядер, не входящих в этот фрагмент, при таком нормальном колебании очень малы. Па этом основана широко используемая в структурно-аналит. исследованиях концепция т. наз. групповых, или характеристических, частот: определенные функц. группы или фрагменты, повторяющиеся в молекулах разл. соед., характеризуются примерно одними и теми же частотами в К. с., по к-рым м.б. установлено их присутствие в молекуле данного в-ва (правда, не всегда с одинаково высокой степенью достоверности). Напр., для карбонильной группы характерна очень интенсивная полоса в ИК спектре поглощения в области ~1700(b50) см-1, относящаяся к валентному колебанию 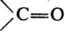 . Отсутствие полос поглощения в данной области спектра доказывает, что в молекуле исследуемого в-ва группы . Отсутствие полос поглощения в данной области спектра доказывает, что в молекуле исследуемого в-ва группы 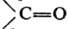 нет. В то же время наличие к.-л. полос в указанной области еще не является однозначным доказательством присутствия в молекуле карбонильной группы, т.к. в этой области могут случайно оказаться частоты других колебаний молекулы. Поэтому структурный анализ и определение конформаций по колебат. частотам функц.
групп должны опираться на неск. характеристич. частот, а предполагаемая структура молекулы должна подтверждаться данными др. методов (см. Структурная химия). Существуют справочники, содержащие многочисл. структурно-спектральные корреляции; имеются также банки данных и соответствующие программы для информационно-поисковых систем и структурно-аналит. исследований с использованием ЭВМ.
Правильной интерпретации К. с. помогает изотопич. замещение атомов, приводящее к изменению колебат. частот. Так, замена водорода на дейтерий приводит к уменьшению частоты валентного колебания X—Н примерно в 1,4 раза. При изотопич. замещении силовые постоянные молекулы Ке сохраняются. Существует ряд изотопич. правил, позволяющих относить наблюдаемые колебат. частоты к тому или иному типу симметрии колебаний, функц. группам и т.д.
Модельные расчеты К. с. (частот и интенсивностей полос) при заданных силовых постоянных, к-рые используют для определения структуры молекул, составляют прямую задачу колебат. спектроскопии. Необходимые для этого силовые постоянные и т. наз. электрооптич. параметры (дипольные моменты связей, компоненты тензора поляризуемости и др.) переносят из исследований близких по структуре молекул или получают решением обратной задачи, заключающейся в определении наборов силовых постоянных и электрооптич. параметров многоатомных молекул по наблюдаемым колебат. частотам, интенсивностям и др. эксперим. данным. Определение наборов фундаментальных частот К. с. необходимо для вычисления колебат. вкладов в термодинамич. ф-ции в-в. Эти данные используются в расчетах хим. равновесий и для моделирования технол. процессов.
К. с. позволяют изучать не только внутримол. динамику, но и межмолекулярные взаимодействия. Из них получают данные о пов-стях потенциальной энергии, внутр. вращении молекул, движениях атомов с большими амплитудами. По К. с. исследуют ассоциацию молекул и структуру комплексов разл. природы. К. с. зависят от агрегатного состояния в-ва, что позволяет получать информацию о структуре разл. конденсир. фаз. Частоты колебат. переходов четко регистрируются для мол. форм с очень малым временем жизни (до 10-11 с), напр. для конформеров при высоте потенциального барьера в неск. кДж/моль. Поэтому К. с. применяют для исследования конформац. изомерии и быстро устанавливающихся равновесий.
Об использовании К. с. для количеств. анализа и др. целей, а также о совр. технике колебат. спектроскопии см. в ст. Инфракрасная спектроскопия, Комбинационного рассеяния спектроскопия. нет. В то же время наличие к.-л. полос в указанной области еще не является однозначным доказательством присутствия в молекуле карбонильной группы, т.к. в этой области могут случайно оказаться частоты других колебаний молекулы. Поэтому структурный анализ и определение конформаций по колебат. частотам функц.
групп должны опираться на неск. характеристич. частот, а предполагаемая структура молекулы должна подтверждаться данными др. методов (см. Структурная химия). Существуют справочники, содержащие многочисл. структурно-спектральные корреляции; имеются также банки данных и соответствующие программы для информационно-поисковых систем и структурно-аналит. исследований с использованием ЭВМ.
Правильной интерпретации К. с. помогает изотопич. замещение атомов, приводящее к изменению колебат. частот. Так, замена водорода на дейтерий приводит к уменьшению частоты валентного колебания X—Н примерно в 1,4 раза. При изотопич. замещении силовые постоянные молекулы Ке сохраняются. Существует ряд изотопич. правил, позволяющих относить наблюдаемые колебат. частоты к тому или иному типу симметрии колебаний, функц. группам и т.д.
Модельные расчеты К. с. (частот и интенсивностей полос) при заданных силовых постоянных, к-рые используют для определения структуры молекул, составляют прямую задачу колебат. спектроскопии. Необходимые для этого силовые постоянные и т. наз. электрооптич. параметры (дипольные моменты связей, компоненты тензора поляризуемости и др.) переносят из исследований близких по структуре молекул или получают решением обратной задачи, заключающейся в определении наборов силовых постоянных и электрооптич. параметров многоатомных молекул по наблюдаемым колебат. частотам, интенсивностям и др. эксперим. данным. Определение наборов фундаментальных частот К. с. необходимо для вычисления колебат. вкладов в термодинамич. ф-ции в-в. Эти данные используются в расчетах хим. равновесий и для моделирования технол. процессов.
К. с. позволяют изучать не только внутримол. динамику, но и межмолекулярные взаимодействия. Из них получают данные о пов-стях потенциальной энергии, внутр. вращении молекул, движениях атомов с большими амплитудами. По К. с. исследуют ассоциацию молекул и структуру комплексов разл. природы. К. с. зависят от агрегатного состояния в-ва, что позволяет получать информацию о структуре разл. конденсир. фаз. Частоты колебат. переходов четко регистрируются для мол. форм с очень малым временем жизни (до 10-11 с), напр. для конформеров при высоте потенциального барьера в неск. кДж/моль. Поэтому К. с. применяют для исследования конформац. изомерии и быстро устанавливающихся равновесий.
Об использовании К. с. для количеств. анализа и др. целей, а также о совр. технике колебат. спектроскопии см. в ст. Инфракрасная спектроскопия, Комбинационного рассеяния спектроскопия.
===
Исп. литература для статьи «КОЛЕБАТЕЛЬНЫЕ СПЕКТРЫ»: Вильсон Е., Дешиус Дж., Кросс П., Теория колебательных спектров молекул, пер. с англ., М., 1960; Свердлов Л. М., Ковнер М. А., Крайнев Е. П., Колебательные спектры многоатомных молекул, М., 1970; Колебания молекул, 2 изд., М., 1972. Ю. А. Пептин.
Страница «КОЛЕБАТЕЛЬНЫЕ СПЕКТРЫ» подготовлена по материалам химической энциклопедии.
|



