|
ПОЛИКОНДЕНСАЦИЯ (от
греч. polys - многочисленный и конденсация), синтез полимеров взаимодействием
би- или полифункцион. мономеров и(или) олигомеров, обычно сопровождающийся выделением
низкомол. продукта (воды, спирта, NH3, галогеноводорода, соответствующих
солей и ДР·)·
Классификация и терминология
П. бифункцион. мономеров
наз. линейной; П., в к-рой участвует хотя бы один мономер, имеющий более двух
функц. групп,-трехмерной (образуются соотв. линейные и сетчатые полимеры). По
типу (и числу) участвующих в р-ции мономеров различают гомополиконденса-цию
(участвует минимально возможное число типов мономеров-о дин или два) и со поликонденсацию.
Важная разновидность П.-полициклоконденсация, при к-рой продукт линейной П.
подвергается внутримол. циклизации (подробнее см. Полициклизация).
П.-ступенчатый процесс,
при к-ром мономеры, взаимодействуя друг с другом, исчерпываются на сравнительно
ранней стадии р-ции, а высокомол. полимер образуется обычно в результате р-ций
ранее образовавшихся олигомеров
и полимерных цепей при глубине превращения функц. групп, близкой к 100%. При
П. часто возможны обратная и разл. обменные р-ции (см. ниже). В зависимости
от вклада этих р-ций различают равновесную (обратимую) П., когда вклад указанных,
особенно обратной, р-ций велик, и неравновесную (необратимую) П., когда эти
р-ции не оказывают практически влияния на процесс образования полимера. К первой
условно относят П. с константой равновесия (Kp больше
103, ко второй-с Крменее 103 (обычно
0,1-10). При неравновесной П. обычно образуются значительно более высокомол.
полимеры (мол. м. 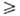 100000),
чем при равновесной (мол. м. 20000-30000). 100000),
чем при равновесной (мол. м. 20000-30000).
Мономеры
В качестве мономеров в
П. используют соед., содержащие в молекуле не менее двух функц. групп. Их можно
разделить на три осн. типа. 1) Мономеры (напр., диамины или дихлор-ангидриды
дикарбоновых к-т), содержащие в молекулах одинаковые функц. группы, не способные
в определенных условиях реагировать между собой. Полимер в этом случае образуется
в результате П. разных мономеров, способных взаимодействовать друг с другом,
напр.:
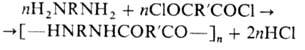
2) Мономеры (напр., гидроксикислоты
или аминокислоты), содержащие разл. функц. группы, к-рые способны реагировать
друг с другом, приводя к образованию полимера, напр.:
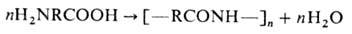
3) Мономеры, содержащие
одинаковые функц. группы, способные реагировать между собой в данных условиях,
напр. гликоли, П. к-рых приводит к образованию простых полиэфиров:
В условиях П. возможны
случаи, когда функц. группы одного или неск. мономеров реагируют как друг с
другом, так и между собой, обусловливая появление разнозвен-ности полимеров.
Примером может служить образование наряду со сложноэфирными простых эфирных
связей (диэтиленгликольных фрагментов) при синтезе полиэтилен-терефталата из
диметилтерефталата и этиленгликоля.
Функциональность и активность
функц. групп мономера зависит от его строения, условий П. и природы реагирующего
с ним мономера. При этом из определенных полифунк-цион. мономеров (мономеры
с числом функц. групп больше двух) получают как линейные, так и сетчатые полимеры.
Так, при П. бис-(о-фенилендиаминов) с биc-(a-дикетонами)
образуются линейные, плавкие и р-римые полифенилхино-ксалины, тогда как при
р-ции этих же тетрафункцион. мономеров с дихлорангидридами дикарбоновых к-т
получаются разветвленные и сетчатые полиамиды. Поэтому различают возможную,
практическую и относительную (определяется как отношение практич. функциональности
к возможной) функциональности мономеров.
Для П. наиб. распространены
мономеры с амино-, кар-бокси-, гидрокси-, меркаптогруппами, из к-рых получают
важнейшие типы поликонденсац. полимеров. В отличие от полимеризации, при к-рой
для получения определенного полимера обычно требуется один мономер, поликонденсац.
полимеры одного типа можно синтезировать из мономеров с самыми разнообразными
функц. группами. Напр., при получении сложных полиэфиров гидроксилсодержащие
мономеры м. б. заменены на галогенсодержащие или сложные эфиры, а вместо карбоновых
к-т можно использовать их хлорангидриды, эфиры, соли, ангидриды и т. п. Естественно,
что при такой замене меняются закономерности и условия П., тип катализа, характер
концевых групп в образующихся макромолекулах и, кроме того, появляются возможности
получения полимеров
с заданным комплексом технол. и эксплуатац. св-в.
Основная и побочные
реакции
В общем виде схема основной
р-ции П.-роста цепи-м. б. представлена след, образом.:
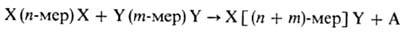
(n и m-любое
целое число, включая единицу, X и Y-исходные функц. группы, А-низкомол. продукт
П.). При этом взаимод. мономеров друг с другом или с образовавшимися олигомерами
и последних между собой подчиняется практически одним и тем же законам.
Поскольку при П. мономеры
исчерпываются уже при невысоких степенях завершенности р-ции, рост цепи высокомол.
полимера происходит преим. в результате многократного соединения между собой
олигомерных или полимерных молекул по концевым функц. группам (принцип многократного
удвоения), при этом число молекул в системе уменьшается (в этом ступенчатый
характер П.). Уменьшается в ходе П. и кол-во исходных функц. групп-реакционных
(активных) центров, хотя в ряде случаев образующиеся при П. связи реагируют
как между собой, так и с исходными реакц. центрами. Росту полимерной цепи при
равновесной П. сопутствует обратная р-ция полимера с выделяющимся низкомол.
продуктом, что ограничивает мол. массу полимера.
При П. функц. группы мономеров,
олигомеров и полимерных цепей расходуются не только на рост цепи, но и на побочные
р-ции (р-ции с примесями или со специально введенными в процесс в-вами, декарбоксилирование
карбоновых к-т и др.), что также лимитирует мол. массу образующегося полимера.
При П. возможны также циклизация и обменные р-ции. Циклизация м. б. внутримолекулярной,
когда кольца образуются при р-ции функц. групп одной молекулы, или межмолекулярной
при взаимод. двух или более молекул одинаковой или разл. природы, напр.:
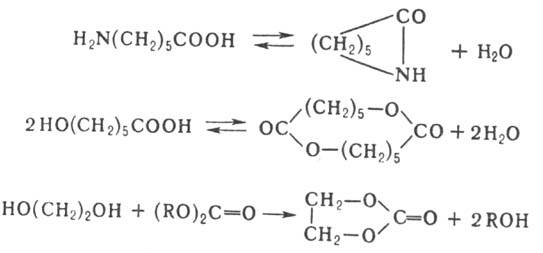
Возможность циклизации
определяется соотношением двух факторов: 1) снижения вероятности образования
цикла по мере увеличения его размера (увеличение энтропии активации); 2) напряженности
цикла, к-рая уменьшается при увеличении его размера вплоть до 6-членного, затем
возрастает при изменении числа членов в цикле от 6 до 9-11, а затем вновь снижается
при переходе к еще большим циклам. В результате совместного действия обоих этих
факторов при получении методом П. разл. полимеров возможно образование больших
циклов (20-40-членных). Циклообразованию способствует проведение р-ции в сильно
разб. р-рах (см. Краун-эфиры).
Обменные р-ции особенно
эффективны при повыш. т-рах П. Их делят на два осн. типа: 1) р-ции обмена образовавшихся
при П. групп (сложноэфирной, амидной или др.) и даже нек-рых циклов (напр.,
имидного) с функц. группами мономеров или примесей (напр., алкоголиз, ацидолиз,
аминолиз); 2) р-ции межцепного обмена между образовавшимися при П. одно- или
разнотипными группами (напр., эфиролиз, амидолиз). Эффективность обменных р-ций
зависит от соотношения скоростей основной и побочных р-ций. Обменные р-ции могут
существенно влиять на мол. массу и MMP поликонденсац. полимера, микроструктуру
сополимера. В ряде
случаев обменные р-ции положены в основу получения поликонденсац. гомо- и сополимеров,
напр, синтез поли-этилентерефталата переэтерификацией диметилтерефталата этиленгликолем.
Ограничение роста полимерной
цепи м. б. обусловлено и чисто физ. причинами, напр. преждевременным выпадением
полимера из реакц. среды в осадок при П. в р-ре (особенно если это сопровождается
его кристаллизацией), однако если выпадающий из р-ра полимер набухает в реакц.
среде, рост цепи часто не прекращается.
Кинетика, катализ, молекулярно-массовое
распределение
Своеобразие П., существенно
отличающее ее от полимеризации, заключается в следующем: исчезновение мономеров
в реакц. среде, как уже отмечалось, наступает задолго до образования полимера
достаточно высокой степени полимеризации, или мол. массы; в большинстве случаев
(при П. в гомог. условиях) получение полимера высокой мол. массы возможно лишь
при очень высокой (близкой к количественной) степени завершенности р-ции (глубине
превращения). Это наглядно иллюстрирует след. зависимость средней степени полимеризации
 полимера
от степени завершенности р-ции (х): полимера
от степени завершенности р-ции (х):  = 1/(1 — x). Поэтому особенно важно изучение кинетики П., имеющей
специфику при протекании в гомо-или гетерогенных условиях.
= 1/(1 — x). Поэтому особенно важно изучение кинетики П., имеющей
специфику при протекании в гомо-или гетерогенных условиях.
Кинетика П., включающей
бесконечное число актов роста цепи, как правило, с трудом поддается количеств.
анализу. Для его упрощения вводят след. допущения (допущения Fлори): 1)
реакц. способности обеих однотипных функц. групп бифункцион. мономера одинаковы;
2) реакц. способность одной функц. группы бифункцион. мономера не зависит от
того, прореагировала или нет др. группа; 3) реакц. способность функц. группы
не зависит от размера молекулы, с к-рой она связана. При этом описание кинетики
П. становится таким же, как кинетики аналогичных р-ций низкомол. соединений
(см. Кинетика химическая). Во многих случаях указанные допущения перестают
быть справедливыми: имеются бифункцион. мономеры, у к-рых однотипные функц.
группы различны по активности или у к-рых реакц. способность второй функц. группы
уменьшается или возрастает после того, как прореагировала первая.
Кинетику П. описывают обычно
до степени завершенности р-ции 0,90-0,95, т.е. когда высокомол. полимер в реакц.
системе практически еще отсутствует. Кинетич. исследование р-ций П., в к-рых
допущения Флори не выполняются, а также заключит. стадий П. (собственно р-ций
поли-мерообразования), когда существ. роль начинают играть диффузионные факторы,
требует применения более сложных кинетич. ур-ний и систем ур-ний, решение к-рых
связано с использованием вычислит. техники.
Кинетическая и др. характеристики
равновесной и неравновесной П. сильно различаются. Равновесная П. характеризуется
малыми скоростями [константа скорости 10 -3 -10 -5 л/(моль·с)],
сравнительно большими значениями энергии активации (80-160 кДж/моль), она м.
б. как экзо-, так и эндотермической. Неравновесная П. обычно протекает с высокой
скоростью [константа скорости до 105 л/(моль·с)], как правило, она
сильно экзотермична и характеризуется низкими значениями энергии активации (8-40
кДж/моль).
Максимально возможная мол.
масса полимера, получаемого равновесной П., определяется термодинамич. факторами
и м. б. определена по ур-нию: 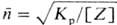 , где [Z]-концентрация низкомол. продукта П. в реакц. системе.
, где [Z]-концентрация низкомол. продукта П. в реакц. системе.
Из этого соотношения следует,
что для получения высокомол. полимеров П. следует проводить в условиях, максимально
благоприятствующих удалению или связыванию низкомол. продукта, напр. в вакууме,
в токе инертного газа, при высоких т-рах. В неравновесной П. мол. масса полимера
не определяется термодинамич. факторами; большое значение для получения высокомол.
полимеров в этом процессе имеет уменьшение вклада побочных р-ций.
Процессы П. можно проводить
как в одну, так и в неск. стадий (см. Полициклизация)., каждая из к-рых
м. б. равновесной
или неравновесной, и в широком диапазоне т-р. В этой связи важно знать не только
константу равновесия всего процесса или отдельных его стадий, но и зависимость
ее от т-ры.
Кинетика П. в гетерог.
системах, когда появляется граница раздела, должна учитывать распределение между
фазами реагентов, их поверхностную активность, разл. процессы массопереноса
и др. факторы. T. обр., кинетика П. (межфазная П.) может определяться не только
хим. взаимодействием функц. групп, но и диффузионными факторами.
Для ускорения П. используют
разл. приемы: активацию функц. групп (напр., замена карбоксильных групп на хлоран-гидридные
или сложноэфирные группы, содержащие остатки сильно кислых фенолов, напр, нитрофенола);
применение активных р-рителей (напр., ДМФА, ДМСО, N, N-диметил-ацетамида, N-метилпирролидона);
введение активирующих агентов (напр., пиридина и трифенилфосфита при П. дикар-боновых
к-т и диаминов); катализ.
Катализаторами служат карбоновые
к-ты, их соли, алко-голяты, третичные амины, сложные эфиры, фосфорные к-ты и
многие др. соед. или их смеси, ускоряющие отдельные стадии П., напр. синтез
полиэтилентерефталата в присут. ацетатов металлов и Sb2O3.
Нек-рые соед., напр. краун-эфиры,
четвертичные аммониевые и фосфониевые соли, применяемые для катализа П., протекающей
в гетерог. условиях, являются катализаторами межфазного переноса, облегчая перенос
реагентов из одной фазы в другую, напр. из водной в органическую. Процессы П.
могут быть также автокаталитическими, напр. синтез полиамидокислот из диангидридов
тетракарбоновых к-т и диаминов, катализируемый полиамидокислотой, и самокатализируемыми,
напр. синтез сложных полиэфиров из ди-карбоновых к-т и диолов, катализируемый
дикарбоновыми к-тами. Катализаторы применяют как в истинно каталитич. кол-вах
(молярные проценты), так и в значительно больших (напр., стехиометрических)
в тех случаях, когда катализатор выполняет также роль акцептора низкомол. продукта
П., напр. П. бисфенолов и дихлорангидридов дикарбоновых к-т в присут. третичных
аминов (акцепторно-каталити-ческая П.).
MMP полимера, получаемого
линейной П., часто описывается ур-нием:
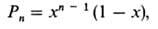
где Pn-молярная
или численная доля n-меров. Это распределение обычно наз. наиб. вероятным
или распределением Флори. См. также Молекулярно-массовое распределение.
Необходимым условием образования
высокомол. полимера при линейной П. является стехиометрич. соотношение реагирующих
функц. групп. При отклонении от этого соотношения обычно образуются более низкомол.
полимеры. Степень полимеризации полимера, синтезируемого при избытке (или недостатке)
одного из мономеров, м. б. определена по ур-нию:
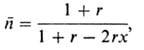
где r-стехиометрич.
разбаланс, т.е. соотношение кол-в функц. групп обоих мономеров. При x 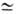 1
указанное ур-ние принимает вид: 1
указанное ур-ние принимает вид:
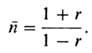
П. при заданном избытке
одного из мономеров-способ регулирования мол. массы полимеров и получения реакци-онноспособных
полимеров или олигомеров (блоков) с определенным типом функц. групп; их затем
используют в синтезе высокомол. полимеров. Так поступают, напр., при синтезе
полиуретанов; вначале из дикарбоновых к-т и избытка гликолей получают сложные
полиэфиры с концевыми группами
ОН, а затем проводят их взаимод. с диизоциана-тами.
Способы проведения поликонденсации
Выбор способа проведения
П. определяется физ.-хим. св-вами исходных в-в и образующихся полимеров, технол.
требованиями, задачами, к-рые ставятся при осуществлении процесса, и т.д. По
т-ре способы проведения П. делят на высокотемпературные и низкотемпературные
(см. табл.), по агрегатному состоянию реакц. системы или фазовому состоянию-на
П. в массе (расплаве), твердой фазе, р-ре, эмульсии (суспензии), двухфазной
системе (межфазная П.). П. в расплаве и твердой фазе происходит при высоких
т-рах, П. в эмульсии и межфазная П.-при низких т-рах, П. в р-ре-при высоких
и низких т-рах. Низкотемпературная П. является преим. неравновесной, высокотемпературная
-преим. равновесной. См. также Межфазная поликонденсация, Поликонденсация
в расплаве, Поликонденсация в растворе.
СРАВНЕНИЕ МЕТОДОВ НИЗКОТЕМПЕРАТУРНОЙ
И ВЫСОКОТЕМПЕРАТУРНОЙ ПОЛИКОНДЕНСАЦИИ
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Стехиометрич. соотношение
|
Часто допустимы
определенные отклонения
|
|
|
|
|
|
|
|
|
|
|
|
Разнообразное;
ограничено требованием реакц. способности
|
Ограничено термостойкостью;
пониж. требования к реакц. способности
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Иногда весьма сложное
(часто автоклавное)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(20-30)· 103,
иногда выше
|
|
|
|
|
|
Ограничено термостойкостью
|
|
|
|
|
|
Вода или летучие
орг. соединения
|
|
|
|
|
|
|
|
Заключение
Процессы П. играют большую
роль в природе и технике. П. лежит в основе образования белков, целлюлозы, крахмала,
нуклеиновых к-т и Др. Первое пром. произ-во синте-тич. полимера - феноло-формальд.
смолы (Л. Бакеланд, 1909), основано на р-циях П. Большой вклад в развитие процессов
П. внесли отечеств. ученые: В. В. Коршак, Г. С. Петров, К. А. Андрианов, амер.
ученые У. Карозерс, П. Флори, П. Морган.
П. широко используют для
получения крупнотоннажных полимеров (сложных полиэфиров, полиамидов, поликарбонатов,
феноло- и мочевино-формальд. смол), нек-рых типов кремнийорг. полимеров, полимеров
со спец. св-вами (гл. обр. тепло- и термостойких - полиимидов, полиарилатов,
полисульфонов, ароматич. простых полиэфиров и полиамидов и др.), к-рые находят
применение в авиац. и космич. технике, микроэлектронике, автомобилестроении
и др. отраслях пром-сти.
===
Исп. литература для статьи «ПОЛИКОНДЕНСАЦИЯ»: Морган П.
У., Поликонденсациойные процессы синтеза полимеров, пер. с англ., Л., 1970;
Коршак В. В., Виноградова С. В., Равновесная поликонденсация, M., 1968; Коршак
В. В., Виноградова С. В., Неравно-весная поликонденсация, М., 1972; Оудиан Дж.,
Основы химии полимеров, пер. с англ., M., 1974; Мономеры для поликонденсации.
под ред. Дж. Стилла и T.У. Кемпбелла, пер. с англ., M., 1976; Коршак В. В.,
Разнозвенность полимеров, M., 1977; Соколов Л. Б., Основы синтеза полимеров
методом поликон-деисации, M., 1979. Я. С. Выгодский.
Страница «ПОЛИКОНДЕНСАЦИЯ» подготовлена по материалам химической энциклопедии.
|



