|
АДСОРБЦИЯ (от лат. ad-на, при и sorbeo-поглощаю), изменение (обычно-повышение)
концентрации в-ва вблизи пов-сти раздела фаз ("поглощение на пов-сти").
В общем случае причина А.-нескомпенсированность межмол. сил вблизи этой
пов-сти, т.е. наличие адсорбц. силового поля. Тело, создающее такое поле,
наз. адсорбентом, в-во, молекулы к-рого могут адсорбироваться,-а д с о
р б т и в о м, уже адсорбиров. в-во-адсорбатом. Процесс, обратный А., наз.
десорбцией.
Природа адсорбц. сил м. б. весьма различной. Если это ван-дер-ваальсовы
силы, то А. наз. физической, если валентные (т.е. А. сопровождается образованием
поверхностных хим. соединений), - химической, или хемосорбцией. Отличит.
черты хемосорбции - необратимость, высокие тепловые эффекты (сотни кДж/моль),
активированный характер. Между физ. и хим. А. существует множество промежут.
случаев (напр., А., обусловленная образованием водородных связей). Возможны
также разл. типы физ. А. наиб. универсально проявление дисперсионных межмол.
сил притяжения, т. к. они приблизительно постоянны для адсорбентов с пов-стью
любой хим. природы (т. наз. неспецифич. А.). Физ. А. может быть вызвана
электростатич. силами (взаимод. между ионами, диполями или квадруполями);
при этом А. определяется хим. природой молекул адсорбтива (т. наз. специфич.
А.). значит. роль при А. играет также геометрия пов-сти раздела: в случае
плоской пов-сти говорят об А. на открытой пов-сти, в случае слабо или сильно
искривленной пов-сти-об А. в порах адсорбента.
В теории А. различают статику (система адсорбент-ад-сорбат находится
в термодинамич. равновесии) и кинетику (равновесия нет).
Статика адсорбции
Термодинамика. Основы термодинамики А. были созданы Дж. Гиббсом
в 70-е гг. 19 в. По Гиббсу, в равновесной двухфазной системе вблизи пов-сти
раздела фаз происходит нек-рое изменение локальных значений всех экстенсивных
св-в (кроме объема). Однако фазы считаются однородными вплоть до нек-рой
геом. пов-сти, разделяющей их. Поэтому значение к.-л. экстенсивного св-ва
для системы в целом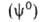 не равно сумме значений этого св-ва в однородных фазах
не равно сумме значений этого св-ва в однородных фазах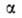 и
и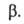 Разность Разность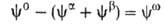 приписывается двухмерной поверхностной фазе, связанной с разделяющей пов-стью.
Т. к. поверхностная фаза не имеет толщины, то V0 =
приписывается двухмерной поверхностной фазе, связанной с разделяющей пов-стью.
Т. к. поверхностная фаза не имеет толщины, то V0 =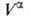 + +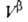 и
и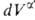 = — = —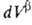 ,
где V- объем. ,
где V- объем.
Изложенные представления позволяют привести фундам. термодинамич. ур-ние
к виду:
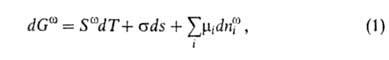
где G-гиббсова своб. энергия, 5-энтропия,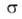 -межфазное
поверхностное натяжение, 5-площадь пов-сти раздела, -межфазное
поверхностное натяжение, 5-площадь пов-сти раздела,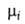 и
иi--соотв. хим. потенциал и число молей i-того
компонента. Индекс и
иi--соотв. хим. потенциал и число молей i-того
компонента. Индекс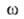 указывает на значение соответствующего св-ва в поверхностной фазе. Преобразование
Лежандра позволяет видоизменить ур-ние (1) для изотермич. условий:
указывает на значение соответствующего св-ва в поверхностной фазе. Преобразование
Лежандра позволяет видоизменить ур-ние (1) для изотермич. условий:
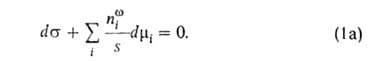
Величина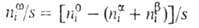 наз. гиббсовой А. и обозначается символом Г, (выражается в моль/см2).
Для двух-компонентной системы:
наз. гиббсовой А. и обозначается символом Г, (выражается в моль/см2).
Для двух-компонентной системы:
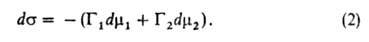
Положение разделяющей пов-сти м. б. выбрано произвольно. В частности,
выбор этого положения может удовлетворять условию Г1 = 0. Такая
пов-сть наз. эквимолекулярной. Для нее вводится обозначение Г2
= Г2(1) Отсюда следует осн. адсорбц. ур-ние Гиббса:

Если адсорбтив совершенно не раств. в одной из двух фаз,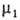 = const, и переход от ур-ния (2) к ур-нию (3) не требует условия Г1
= 0. Т. обр., гиббсова А.-это избыток данного компонента в реальной
двухфазной системе по сравнению с такой системой, в к-рой обе фазы были
бы строго однородны вплоть до разделяющей пов-сти. Кроме гиббсовых избыточных
величин А., в ее теории большую роль играет А., понимаемая как полное содержание
компонента i в пространстве W, в к-ром проявляются адсорбц.
силы. Обозначая полное содержание через а и считая, что компонент
i
совершенно не раств. в одной из объемных фаз, имеем:
= const, и переход от ур-ния (2) к ур-нию (3) не требует условия Г1
= 0. Т. обр., гиббсова А.-это избыток данного компонента в реальной
двухфазной системе по сравнению с такой системой, в к-рой обе фазы были
бы строго однородны вплоть до разделяющей пов-сти. Кроме гиббсовых избыточных
величин А., в ее теории большую роль играет А., понимаемая как полное содержание
компонента i в пространстве W, в к-ром проявляются адсорбц.
силы. Обозначая полное содержание через а и считая, что компонент
i
совершенно не раств. в одной из объемных фаз, имеем:

где ci-концентрация i-того компонента в объемной
фазе. При малых сi:
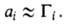
А. может происходить на любой пов-сти раздела между двумя любыми фазами,
в частности на пов-сти раздела флюид-флюид (жидкость - газ, жидкость -
жидкость) или твердое тело-флюид (твердое - газ, твердое-жидкость). В системах
флюид-флюид можно измерить а как ф-цию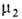 и экспериментально определить Г2(1) по уравнению
(3). Во втором случае ппя определения Г2(1) измеряют
любым методом ni0,
и экспериментально определить Г2(1) по уравнению
(3). Во втором случае ппя определения Г2(1) измеряют
любым методом ni0,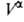 , ,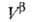 и концентрации i-того компонента в этих объемах. Отсюда вычисляют Гi(1)
Этот метод наз. объемным (или волюмометрич.). При весовом (гравиметрии.)
методе непосредственно определяют кол-во i-того компонента на пов-сти
раздела.
и концентрации i-того компонента в этих объемах. Отсюда вычисляют Гi(1)
Этот метод наз. объемным (или волюмометрич.). При весовом (гравиметрии.)
методе непосредственно определяют кол-во i-того компонента на пов-сти
раздела.
Изотерма адсорбции. В равновесной адсорбц. системе параметры,
определяющие равновесие,-это ai парциальные давления
р
(или сi) и т-ра Т. Они связаны т. наз. термич.
ур-нием:
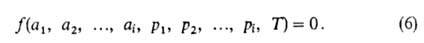
При А. индивидуального адсорбтива (i=1) это ур-ние принимает
вид:

Три частных случая термич. ур-ния (когда Т, р или a-константы)
играют особую роль в теории А.:
а=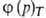 -ур-ние
изотермы А., -ур-ние
изотермы А.,
Т=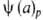 -ур-ние
изобары А., -ур-ние
изобары А.,
Р-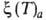 -ур-ние
изостеры А. -ур-ние
изостеры А.
Конкретный вид ф-ций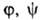 и
и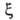 определяется
особенностями рассматриваемой системы. Если одна из них, напр. определяется
особенностями рассматриваемой системы. Если одна из них, напр.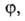 известна для любого значения Т= const, то, очевидно, становятся
известными и две другие. При этом не обязательно, чтобы был известен аналит.
вид зависимостей. Они м. б. заданы эмпирически в виде набора значений а,
р и Т.
известна для любого значения Т= const, то, очевидно, становятся
известными и две другие. При этом не обязательно, чтобы был известен аналит.
вид зависимостей. Они м. б. заданы эмпирически в виде набора значений а,
р и Т.
В теории А. обычно решается вопрос о виде ф-ции а =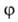 (р)г,
т.е. об ур-нии изотермы А. Эта проблема связана с тепловыми эффектами,
сопровождающими А. При расчете изменения значений осн. термодинамич. ф-ций
в случае перехода dn молей адсорбтива из объемной фазы в поверхностную
в равновесной системе при р = const возможны два случая: в первом
учитывается только превращ. адсорбтива в адсорбат, поскольку адсорбент
при А. термодинамически неизменен и его роль-служить источником адсорбц.
поля; во втором учитывается и изменение адсорбента. (р)г,
т.е. об ур-нии изотермы А. Эта проблема связана с тепловыми эффектами,
сопровождающими А. При расчете изменения значений осн. термодинамич. ф-ций
в случае перехода dn молей адсорбтива из объемной фазы в поверхностную
в равновесной системе при р = const возможны два случая: в первом
учитывается только превращ. адсорбтива в адсорбат, поскольку адсорбент
при А. термодинамически неизменен и его роль-служить источником адсорбц.
поля; во втором учитывается и изменение адсорбента.
Т.к. система равновесна, то хим. потенциалы адсорбата и адсорбтива
одинаковы; энтропия адсорбата вследствие уменьшения подвижности молекул
при А. меньше энтропии адсорбтива. Поэтому при инертном адсорбенте энтальпия
всегда отрицательна, т.е. А. экзотермична. Учет изменения энтропии адсорбента
может изменить этот вывод. Напр., при сорбции полимерами в-в, в к-рых полимер
набухает, энтропия последнего (из-за увеличения подвижности макромолекул)
может столь сильно возрасти, что А. становится эндотермической. В дальнейшем
в статье рассматривается только экзотермич. А.
Различают интегральную, дифференц., изостерич. и среднюю теплоты А.
Интегральная теплота Q равна убыли энтальпии (при V= const
-внутр. энергии) при изменении А. от a1 до а2(в
частном случае м.б. а1=0): Q= -(Н2 - Н1)
Эту величину относят обычно к массе адсорбента и выражают в Дж/кг.
Дифференц. теплота q (Дж/моль) равна убыли энтальпии dH при
изменении а на da. Ее выражают отношением q = — (dH/da).
Очевидно,
что
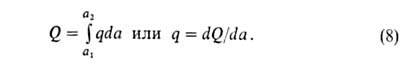
Изостерич. теплоту qst принимают равной:
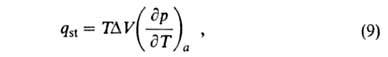
где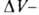 разность
мольных объемов адсорбата и адсорбтива. Можно показать, что разность
мольных объемов адсорбата и адсорбтива. Можно показать, что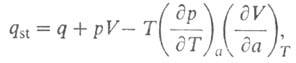 для идеального газового адсоритива:
для идеального газового адсоритива:

Смысл введения qsi в том, что для ее определения не
требуется калориметрич. данных (таких, как Q и q)и она м.
б. вычислена по ур-нию (9) по результатам измерения А. Вводят также среднюю
теплоту Q (Дж/моль):
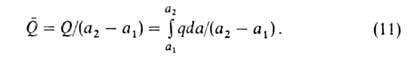
С ростом а параметр Q всегда возрастает, a q может
уменьшаться, увеличиваться или оставаться неизменной. С ростом а при
неоднородной пов-сти А. происходит на все менее активных участках, что
приводит к уменьшению q. Однако при этом уменьшаются средние расстояния
между адсорбиров. молекулами, вследствие чего увеличиваются силы притяжения
между ними, и q возрастает. Соотношение между двумя упомянутыми
эффектами определяет ход зависимости q =f(a). При очень больших
а
начинают преобладать силы отталкивания и в этой области q всегда
снижается с ростом а.
При очень малых заполнениях пов-сти ур-ние изотермы А. имеет вид ур-ния
Генри:
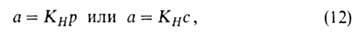
где КH-коэф. Генри. Действительно, при очень малых а адсорбц.
слой подобен двухмерному идеальному газу, поэтому его ур-ние состояния
имеет вид: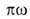 =
RT,
где =
RT,
где -двухмерное давление,
-двухмерное давление,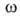 -площадь,
занимаемая одним молем в-ва. Отсюда, учитывая, что -площадь,
занимаемая одним молем в-ва. Отсюда, учитывая, что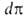 =
— =
—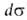 , и используя
ур-ние (3), получаем ур-ние (12). Ур-ние Генри требует, чтобы q было
постоянным. При больших заполнениях это ур-ние перестает выполняться. Поэтому
Г. Фрейндлих (1906) предложил описывать изотермы А. след. эмпирич. ур-нием
(ур-ние Фрёйндлиха): , и используя
ур-ние (3), получаем ур-ние (12). Ур-ние Генри требует, чтобы q было
постоянным. При больших заполнениях это ур-ние перестает выполняться. Поэтому
Г. Фрейндлих (1906) предложил описывать изотермы А. след. эмпирич. ур-нием
(ур-ние Фрёйндлиха):

где k и п - константы. Этим ур-нием часто пользуются как
интерполяц. ф-лой, хотя оно при малых р не переходит в ур-ние (12),
а при очень больших р приводит к несогласующемуся с опытом неогранич.
возрастанию а.
Строгая теория изотермы А. была создана И. Ленгмю-ром (1914-18). В основу
теории положена след. модель: 1) пов-сть адсорбента представляет собой
набор энергетически одинаковых активных центров, на к-рых адсорбируются
(локализуются) молекулы адсорбтива; 2) на одном центре адсорбируется только
одна молекула, т.е. при А. образуется только один адсорбц. слой (монослой);
3) А. на данном центре не влияет на А. на др. центрах, т. е. взаимод. адсорбированных
молекул можно пренебречь.
Модель Ленгмюра наз. локализованной мономолекулярной А. на однородной
пов-сти. Ур-ние изотермы А., соответствующее этой модели, м.б. получено
при помощи разл. методов (молекулярно-кинетич., термодинамич., ста-тистико-термодинамич.).
Так, адсорбц. равновесие можно выразить след. схемой:
Молекула Своб. Адсорбц. в газовой + адсорбц.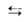 комплекс фазе центр (занятый центр)
комплекс фазе центр (занятый центр)
Концентрация молекул в газе пропорциональна р, концентрация своб. центров-величине
(ат
— а), где ат- полное число центров, а-число занятых
центров, концентрация адсорбц. комплексов-величине а. Следовательно, константа
равновесия равна: Кр = р(ат — а)/а. Отсюда
получаем ур-ние Ленгмюра:
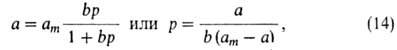
где b-т. наз. адсорбц. коэф., равный Кр-1.
В
области очень малых давлений bр " 1 и a = (amb)p,
что
отвечает ур-нию Генри, в к-ром КH = amb.
В
области очень больших давлений bр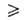 1
и а 1
и а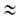 ат;
при
этом А. перестает зависеть от давления. Константа равновесия b-1связана
со стандартным значением изобарного потенциала р-ции: ат;
при
этом А. перестает зависеть от давления. Константа равновесия b-1связана
со стандартным значением изобарного потенциала р-ции:
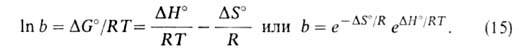
Модель Ленгмюра требует, чтобы дифференц. теплота и энтропия А. не зависели
от степени заполнения пов-сти.
Ур-ние (14)-строгое выражение, соответствующее модели Ленгмюра, однако
оно редко оправдывается на опыте, поскольку сама модель идеализирована.
Учение об А. с 20-х гг. 20 в. в значит. степени строилось на основе ослабления
или исключения того или иного допущения Ленгмюра.
Уже Ленгмюр предложил способ описания А. на неоднородной пов-сти (т.е.
при допущении, что не все центры оди наковы). Объединяя одинаковые центры
в группы и полагая, что к каждой группе применимо ур-ние (14), можно считать,
что А. на всей пов-сти выражается суммой членов ур-ния (14):
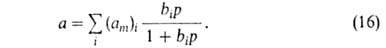
Полагая, что число адсорбц. центров м.б. описано непрерывной ф-цией
распределения по значениям своб. энергии, Я.Б.Зельдович получил из ф-лы
(16) для экспоненциальной ф-ции ур-ние типа (13).
А. на неоднородных пов-стях-большая глава теории А. Ее осн. задача-решение
интегрального ур-ния:
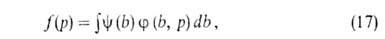
где f(р)- т. наз. эмпирич. изотерма А.,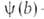 -та или иная ф-ция распределения числа центров по значениям своб. энергии,
-та или иная ф-ция распределения числа центров по значениям своб. энергии,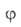 (b,
р)- локальная изотерма А., в кач-ве к-рой обычно принимают изотерму
Ленгмюра. (b,
р)- локальная изотерма А., в кач-ве к-рой обычно принимают изотерму
Ленгмюра.
Много попыток сделано в направлении отказа от второго допущения Ленгмюра.
На этом пути особое значение приобрела теория полимолекулярной А., предложенная
С. Брунауэром, П. Эмметом и Э. Теллером (теория БЭТ). Теория постулирует,
что при т-ре ниже критической каждая молекула, адсорбированная в первом
слое (теплота адсорбции qi,), является центром для молекул,
образующих второй слой, и т.д. При этом считается, что теплота А. во всех
слоях, кроме первого, равна теплоте конденсации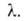 Такая модель приводит к ур-нию:
Такая модель приводит к ур-нию:
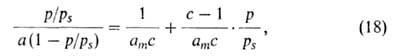
где с = ехр[(q1 -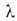 )/RT].
Ур-ние (18) в координатах a, p/ps соответствует S-образной
кривой. В координатах p/ps, )/RT].
Ур-ние (18) в координатах a, p/ps соответствует S-образной
кривой. В координатах p/ps,
изотерма А. по ур-нию (18) должна быть линейной.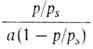 Наклон этой прямой (обычно в интервале 0,05 < p/ps <
< 0,30) и отрезок, отсекаемый ею на оси ординат, дают значения соотв.
ат
и с. Широкое распространение теории БЭТ связано с тем, что ее
авторы, фактически считая А. нелокализованной, отождествляют константу
ат не с числом дискретных адсорбц. центров, а с числом
молекул адсорба-та в первом слое при плотнейшей упаковке (при
р =
ps). Поэтому, вводя представление о площади
Наклон этой прямой (обычно в интервале 0,05 < p/ps <
< 0,30) и отрезок, отсекаемый ею на оси ординат, дают значения соотв.
ат
и с. Широкое распространение теории БЭТ связано с тем, что ее
авторы, фактически считая А. нелокализованной, отождествляют константу
ат не с числом дискретных адсорбц. центров, а с числом
молекул адсорба-та в первом слое при плотнейшей упаковке (при
р =
ps). Поэтому, вводя представление о площади занимаемой одной молекулой в этом слое, принимают:
занимаемой одной молекулой в этом слое, принимают:

где s- площадь пов-сти адсорбата. Как правило, для этого измеряют
изотерму А. азота и принимают, что для его молекулы =
0,162нм2. Часто выполняемый аналогичный расчет s по модели
Ленгмюра не корректен, т.к. этот метод, очевидно, применим только к нелокализованной
А. =
0,162нм2. Часто выполняемый аналогичный расчет s по модели
Ленгмюра не корректен, т.к. этот метод, очевидно, применим только к нелокализованной
А.
В теорию полимолекулярной А. большой вклад внес Я. де Бур, экспериментально
показавший, что зависимость среднего числа слоев (свыше первого) на всех
пов-стях, близких по хим. природе, от p/ps выражается
универсальной кривой (т. наз. t-кривой). Это также позволяет оценивать
площади пов-сти адсорбтивов.
Предпринимались попытки учесть в модели Ленгмюра также взаимод. между
адсорбиров. молекулами. Так, Т. Хилл и Я. де Бур, считая, что ур-ние состояния
адсорбц. слоя есть двухмерный аналог ур-ния Ван-дер-Ваальса, получили след.
ур-ние изотермы А.:
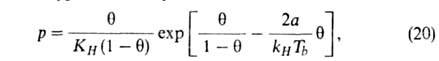
где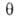 =
а/ат,
а и b-константы ур-ния Ван-дер-Ваальса. Р. Фаулер и Э. Гуггенгейм,
учтя взаимод. адсорбиров. молекул, вывели ур-ние: =
а/ат,
а и b-константы ур-ния Ван-дер-Ваальса. Р. Фаулер и Э. Гуггенгейм,
учтя взаимод. адсорбиров. молекул, вывели ур-ние:
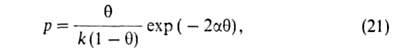
где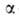 -константа,
связанная с парным взаимодействием молекул. -константа,
связанная с парным взаимодействием молекул.
Существует еще один механизм, приводящий к дополнит. А. адсорбтивов
ниже их критич. т-ры на пористых адсорбентах при сравнительно высоких значениях
p/ps.
Это - капиллярная конденсация. Если в поре образовался вогнутый мениск
адсорбата, то в ней начинается конденсация при p/ps<1.
Согласно
ур-нию Кельвина:
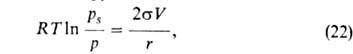
где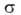 -поверхностное
натяжение адсорбата, V-его мольный объем, r-радиус кривизны мениска.
Капиллярная конденсация приводит к резкому подъему изотермы А. При этом
часто (но не всегда) наблюдается т. наз. адсорбц. гистерезис, т.е. несовпадение
адсорбц. и десорбц. ветвей изотермы. Как правило, это связано с тем, что
формы менисков при А. и десорбции не совпадают. -поверхностное
натяжение адсорбата, V-его мольный объем, r-радиус кривизны мениска.
Капиллярная конденсация приводит к резкому подъему изотермы А. При этом
часто (но не всегда) наблюдается т. наз. адсорбц. гистерезис, т.е. несовпадение
адсорбц. и десорбц. ветвей изотермы. Как правило, это связано с тем, что
формы менисков при А. и десорбции не совпадают.
Капиллярную конденсацию используют для определения размеров пор адсорбента.
По ур-нию (22) для каждого значения p/ps вычисляют радиус
кривизны мениска. Из него, учитывая толщину адсорбц. слоя (напр., по t-кривой),
форму переходной области от слоя к мениску и зависимость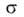 от кривизны при очень малых r, находят линейный размер (эффективный
радиус ref) пор, заполняемых при данном p/ps.
Объем
таких пор определяют по приросту А. в этой точке изотермы. Используя полученные
данные, строят кривую распределения объема пор по их радиусам. Метод применим
при ref
от кривизны при очень малых r, находят линейный размер (эффективный
радиус ref) пор, заполняемых при данном p/ps.
Объем
таких пор определяют по приросту А. в этой точке изотермы. Используя полученные
данные, строят кривую распределения объема пор по их радиусам. Метод применим
при ref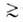 1,5
нм. Обычно расчет ведут по десорбц. ветви изотермы, но более строгая совр.
теория требует для построения кривой учета обеих ветвей. 1,5
нм. Обычно расчет ведут по десорбц. ветви изотермы, но более строгая совр.
теория требует для построения кривой учета обеих ветвей.
Потенциальная теория адсорбции и теория объемного заполнения микропор.
Модель
А., принципиально отличную от ленгмюровской, предложил в 1914 М. Поляки.
Согласно этой модели, вблизи пов-сти адсорбента существует потенциальное
адсорбц. силовое поле, убывающее с расстоянием от пов-сти. Вследствие этого
давление адсорбтива, равное вдали от пов-сти р, вблизи нее возрастает и
на нек-ром расстоянии достигает значения ps, при к-ром адсорбтив
конденсируется. Объем слоя между пов-стыо раздела и геом. местом точек,
где р = ps, заполнен жидкостью, к-рой приписываются нормальные
значения физ. св-в объемной жидкости. Обратимая изотермич. работа е адсорбц.
сил, определяемая по ур-нию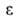 =
RTlnp/ps, наз. адсорбц. потенциалом, а вся концепция-потенциальной
теорией А. При заданной величине объема V адсорбц. слоя потенциал =
RTlnp/ps, наз. адсорбц. потенциалом, а вся концепция-потенциальной
теорией А. При заданной величине объема V адсорбц. слоя потенциал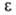 не
зависит от т-ры (вследствие независимости дисперсионных сил от т-ры). Такая
температурная инвариантность дает возможность пересчитывать А. с одной
т-ры на другую, хотя ур-ния изотермы А. на основе излагаемой теории вывести
не удавалось. Модель Поляни широко и успешно применялась мн. авторами,
однако она содержала два очень уязвимых положения: 1) допущение о том,
что тончайшая адсорбц. пленка имеет нормальные значения физ. св-в объемной
жидкости (это допущение не подтверждалось опытами); 2) температурная инвариантность
ф-ции не
зависит от т-ры (вследствие независимости дисперсионных сил от т-ры). Такая
температурная инвариантность дает возможность пересчитывать А. с одной
т-ры на другую, хотя ур-ния изотермы А. на основе излагаемой теории вывести
не удавалось. Модель Поляни широко и успешно применялась мн. авторами,
однако она содержала два очень уязвимых положения: 1) допущение о том,
что тончайшая адсорбц. пленка имеет нормальные значения физ. св-в объемной
жидкости (это допущение не подтверждалось опытами); 2) температурная инвариантность
ф-ции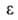 =f(V),
лежащая
в основе теории, приближенно подтверждалась опытом только для очень тонкопористых
адсорбентов. =f(V),
лежащая
в основе теории, приближенно подтверждалась опытом только для очень тонкопористых
адсорбентов.
Используя потенциальную теорию, М.М. Дубинин предложил и разработал
теорию объемного заполнения микро-пор (ТОЗМ). Было постулировано, что эта
теория применима только к микропористым адсорбентам. Особенность таких
адсорбентов, в к-рых линейные размеры пор r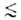 1
нм, состоит в том, что весь объем их пор "заполнен" адсорбц. полем. Поэтому
при А. они заполняются не послойно, а объемно. Величина 1
нм, состоит в том, что весь объем их пор "заполнен" адсорбц. полем. Поэтому
при А. они заполняются не послойно, а объемно. Величина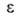 в рассматриваемом случае - это не адсорбц. потенциал, а с точностью до
знака хим. потенциал адсорбата, отсчитываемый от уровня хим. потенциала
нормальной жидкости при той же т-ре. Вся совокупность пор адсорбентов разделяется
на три класса: микропоры (r
в рассматриваемом случае - это не адсорбц. потенциал, а с точностью до
знака хим. потенциал адсорбата, отсчитываемый от уровня хим. потенциала
нормальной жидкости при той же т-ре. Вся совокупность пор адсорбентов разделяется
на три класса: микропоры (r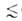 0,6
нм), мезопоры (0,6 нм 0,6
нм), мезопоры (0,6 нм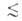 r r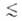 20
нм) и макропоры (r 20
нм) и макропоры (r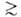 20
нм). А. в микропорах происходит по схеме ТОЗМ, т.е. объемно, в мезопорах-по
механизму послойного заполнения, завершаемого капиллярной конденсацией.
Макропоры при адсорбц. равновесии никакой роли не играют. 20
нм). А. в микропорах происходит по схеме ТОЗМ, т.е. объемно, в мезопорах-по
механизму послойного заполнения, завершаемого капиллярной конденсацией.
Макропоры при адсорбц. равновесии никакой роли не играют.
Введя представление о ф-ции распределения объемов пор по значениям хим.
потенциала адсорбата в них, М.М. Дубинин и Л. В. Радушкевич получили ур-ние
изотермы адсорбции ТОЗМ, к-рое обычно записывают в след. форме:
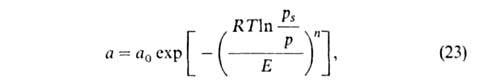
где п, Е и а0-параметры (а0 = а при
р
= ps). Температурная зависимость a0:
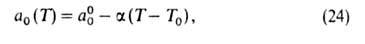
где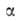 =
-(da0/dT);
a00 = a0 при Т= Т0.
Параметры п и Е практически не зависят от т-ры. В большинстве
случаев п = 2. Лишь для случаев, когда начальные теплоты А. очень
велики, п > 2. Для пересчета изотерм А. с одного адсорбтива на другой
приближенно допускают, что E1/E2 =
-(da0/dT);
a00 = a0 при Т= Т0.
Параметры п и Е практически не зависят от т-ры. В большинстве
случаев п = 2. Лишь для случаев, когда начальные теплоты А. очень
велики, п > 2. Для пересчета изотерм А. с одного адсорбтива на другой
приближенно допускают, что E1/E2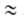 P1/P= P1/P=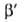 и
что a01/a02 и
что a01/a02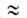 V1/V2,где
Pi-парахор, Vi- мольный объем адсорбтива. V1/V2,где
Pi-парахор, Vi- мольный объем адсорбтива.
Каждый микропористый адсорбент характеризуется по ТОЗМ двумя параметрами:
W-объемом
микропор (W0 = = a0V0)и E0-характеристич.
энергией; W0 и E0 относят к стандартному адсорбтиву,
обычно к бензолу.
Пользуясь представлением, что в реальном адсорбенте имеются поры разных
размеров, и вводя распределение значений Е с дисперсией, равной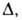 Ф. Стекли предложил обобщение ур-ния (23), названное ур-нием Дубинина-Стёкли:
Ф. Стекли предложил обобщение ур-ния (23), названное ур-нием Дубинина-Стёкли:
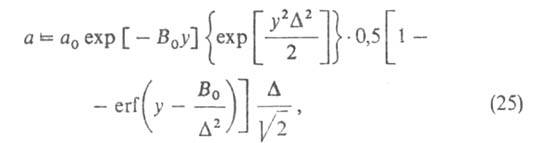
где B0 - константа, связанная с E в ур-нии
(23), а у=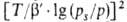 .
T.к. в адсорбц. технике наиб. распространение получили именно микропористые
адсорбенты (активные угли, цеолиты, тонкопористые ксерогели), ТОЗМ применяется
не только в физ.-хим. исследованиях, но и в инженерных расчетах. .
T.к. в адсорбц. технике наиб. распространение получили именно микропористые
адсорбенты (активные угли, цеолиты, тонкопористые ксерогели), ТОЗМ применяется
не только в физ.-хим. исследованиях, но и в инженерных расчетах.
Адсорбция газовых и жидких смесей. Па практике всегда имеют дело не
с индивидуальным адсорбтивом, а со смесью газов или с жидкими р-рами. Поэтому
требуется обобщение теории А. на случай многокомпонентного адсорбтива.
В принципе можно исходить из любой модели А. и распространить ее на этот
случай. При А. газовой смеси это достигается не только большим усложнением
ур-ний, но и введением в них дополнит. эмпирич. параметров, связанных или
с взаимод. разнородных молекул или, в более общем виде, с влиянием одних
в-в на коэф. активности других. Только модель Ленгмюра позволяет получить
ур-ние изотермы А. смеси без параметров, не входящих в ур-ния для А. индивидуальных
в-в. Для этого достаточно учесть, что при адсорбции k-того компонента из
смеси i компонентов часть адсорбц. центров м.б. занята др. молекулами.
Поэтому:
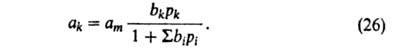
В случае А. жидких р-ров независимо от их концентрации вся пов-сть адсорбента
заполнена. Вследствие этого А. молекулы k-того компонента сопровождается
вытеснением не-к-рого числа молекул остальных компонентов, т. е. А. носит
конкурентный характер.
Различают молекулярную и ионную А. р-ров. Первая происходит при А. р-ров
неэлектролитов, вторая-р-ров электролитов. Молекулярная А., как правило,
выражается избыточными величинами. Конкурентный характер А. обусловливает
то, что величина а м.б. как положительной, так и отрицательной.
Выражая А. i-того компонента как ф-цию его мольной доли в р-ре хi-,
имеем, что Гi = О при хi = 0 и хi
= 1 (возможным изменением объема в-ва в адсорбц. слое пренебрегают). Поэтому
изотерма А. имеет один или неск. экстремумов.
Ур-ние изотермы А. бинарных р-ров неэлектролитов, надежно обоснованное
термодинамически, имеет вид:
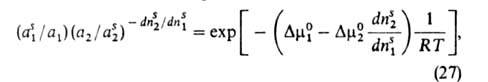
где индекс s указывает на адсорбц. фазу, — (dns2/dns1)показывает,
сколько молей второго компонента вытесняется одним молем первого,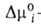 -разность
слагаемых (стандартных частей) хим. потенциала, зависящих только от т-ры. -разность
слагаемых (стандартных частей) хим. потенциала, зависящих только от т-ры.
Осн. проблема использования этого и ряда др. уравнений изотермы А.-выяснение
зависимости коэф. активности компонентов в адсорбц. слое от его состава.
Важнейший вопрос при применении А. для разделения или очистки в-в-подбор
селективного адсорбента по отношению к данному компоненту р-ра.
Ионная А., как правило, не носит эквивалентного характера. На пов-сти
из р-ра электролита адсорбируются преим. катионы или анионы. Благодаря
электрич. (кулонов-ским) силам на пов-сти образуется двойной электрический
слой.
Если в состав адсорбента входят ионы или поверхностные функц. группы,
способные в данном р-рителе к ионизации, то между адсорбентом и р-ром электролита
происходит ионный обмен. Адсорбент в этом случае наз. ионитом.
Кинетика адсорбции
А., как и любой реальный процесс, происходит во времени. Поэтому полная
теория А. должна содержать раздел о кинетике А. Элементарный акт А. осуществляется
практически мгновенно (исключение-хемосорбция). Поэтому временные зависимости
А. определяются в осн. механизмом диффузии, т. е. подвода адсорбтива к
месту А. Если А. на открытой пов-сти не мгновенна, такой процесс происходит
во внешнедиффузионной области; при этом законы диффузии не специфичны для
А. В случае же пористых адсорбентов, кроме внеш. диффузии, важную роль
начинает играть внутр. диффузия, т.е. перенос адсорбтива в порах адсорбента
при наличии в них градиента концентрации. Механизм такого переноса может
зависеть от концентрации адсорбтива и размеров пор.
Различают молекулярную, кнудсеновскую и поверхностную (фольмеровскую)
диффузию. Молекулярная диффузия осуществляется, если длина своб. пробега
молекул в порах меньше размера пор, кнудсеновская-если эта длина превышает
размер пор. При поверхностной диффузии молекулы перемещаются по пов-сти
адсорбента без перехода в объемную фазу. Однако значения коэф. диффузии
не одинаковы для разных механизмов диффузии. Во мн. случаях экспериментально
не удается установить, как именно происходит диффузия, и поэтому вводят
т. наз. эффективный коэф. диффузии, описывающий процесс в целом.
Осн. эксперим. материалом о кинетике А. служит т. наз. кинетич. кривая,
т.е. ф-ция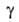 =
а/аравн=f(t) где =
а/аравн=f(t) где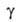 -относительная
А., равная отношению текущего значения адсорбции а к aравн
-
её значению при времени t -относительная
А., равная отношению текущего значения адсорбции а к aравн
-
её значению при времени t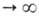 .
Для
истолкования кинетич. кривой в простейшем случае предполагают, что зерно
адсорбента имеет совершенно однородную по объему пористую структуру (эту
модель наз. квазигомогенной). значит. усовершенствование квазигомогенной
модели-представление о том, что каждое зерно содержит области с более крупными
и более тонкими порами. Диффузия в таком зерне описывается двумя разл.
коэффициентами. .
Для
истолкования кинетич. кривой в простейшем случае предполагают, что зерно
адсорбента имеет совершенно однородную по объему пористую структуру (эту
модель наз. квазигомогенной). значит. усовершенствование квазигомогенной
модели-представление о том, что каждое зерно содержит области с более крупными
и более тонкими порами. Диффузия в таком зерне описывается двумя разл.
коэффициентами.
В случае открытой пов-сти, принимая модель Ленгмюра, легко получить
кинетич. ур-ние А. Скорость приближения к равновесию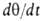 представляет собой разность скоростей А. и десорбции. Считая, как обычно
в кинетике, что скорости процессов пропорциональны концентрациям реагирующих
в-в, имеем:
представляет собой разность скоростей А. и десорбции. Считая, как обычно
в кинетике, что скорости процессов пропорциональны концентрациям реагирующих
в-в, имеем:
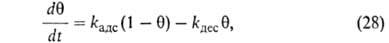
где kадс и kдес- константы скорости соотв. А.
и десорбции. Давление в газовой фазе считается постоянным. При интегрировании
этого ур-ния от t = 0 до любого значения t получим:
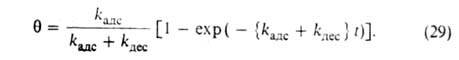
Отсюда при f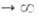 имеем:
имеем: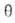 = =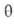 равн.
Поэтому окончательно имеем: равн.
Поэтому окончательно имеем:
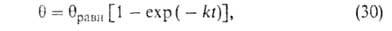
где k = kадс + kдес.
Влияние т-ры на скорость А. выражается ур-нием, аналогичным ур-нию Аррениуса.
С увеличением т-ры kадс экспоненциально возрастает. Т.к. диффузия
в порах адсорбента связана с преодолением активац. барьеров, температурные
зависимости kадс и kдес не одинаковы.
Знание скоростей диффузии важно не только для теории А., но и для расчета
пром. адсорбц. процессов. При этом обычно имеют дело не с отдельными зернами
адсорбента, а с их слоями. Кинетика процесса в слое выражается очень сложными
зависимостями. В каждой точке слоя в данный момент времени величина А.
определяется не только видом ур-ния изотермы А. и закономерностями кинетики
процесса, но также аэро- или гидродинамич. условиями обтекания зерен газовым
или жидкостным потоком. Кинетика процесса в слое адсорбента в отличие от
кинетики в отдельном зерне наз. динамикой А., общая схема решения задач
к-рой такова: составляется система дифференц. ур-ний в частных производных,
учитывающая характеристики слоя, изотерму А., диффузионные характеристики
(коэф. диффузии, виды переноса массы по слою и внутри зерен), аэро- и гидродинамич.
особенности потока. Задаются начальные и краевые условия. Решение этой
системы ур-ний в принципе приводит к значениям величин А. в данный момент
времени в данной точке слоя. Как правило, аналитич. решение удается получить
только для простейших случаев, поэтому такая задача решается численно с
помощью ЭВМ.
При опытном изучении динамики А. через слой адсорбента пропускают газовый
или жидкостный поток с заданными характеристиками и исследуют состав выходящего
потока как ф-цию времени. Появление поглощаемого в-ва за слоем наз. проскоком,
а время до проскока — временем защитного действия. Зависимость концентрации
данного компонента за слоем от времени наз. выходной кривой. Эти кривые
служат осн. эксперим. материалом, позволяющим судить о закономерностях
динамики А.
Аппаратурное оформление адсорбционных процессов
Существует множество технол. приемов проведения адсорбц. процессов.
Широко распространены циклич. (перио-дич.) установки с неподвижным слоем
адсорбента, осн. узел к-рых - один или неск. адсорберов, выполненных в
виде полых колонн, заполняемых гранулированным адсорбентом. Газовый (или
жидкостной) поток, содержащий адсорбируемые компоненты, пропускается через
слой адсорбента до проскока. После этого адсорбент в адсорбере регенерируют,
а газовый поток направляют в др. адсорбер. Регенерация адсорбента включает
ряд стадий, из к-рых ос новная-десорбция, т.е. выделение ранее поглощенного
в-ва из адсорбента. Десорбцию проводят нагреванием, сбросом давления в
газовой фазе, вытеснением (напр., острым водяным паром) или комбинацией
этих методов. Т. к. времена А. и регенерации не совпадают, подбирают такое
число одновременно работающих и регенерируемых адсорберов, чтобы в целом
процесс шел непрерывно.
По техн. и экономич. соображениям регенерацию не доводят до конца. Поэтому
рабочая емкость адсорбента равна разности между максимально достигаемой
в данных условиях А. и кол-вом адсорбата, остающегося в адсорбенте после
регенерации. Вследствие этого изотермы А., соответствующие процессу в адсорбере,
не должны быть слишком крутыми.
В описанной схеме возможны два варианта: 1) целевой продукт адсорбируется
из газового потока практически полностью, и тогда он содержится в десорбате,
откуда его извлекают тем или иным способом; 2) целевой продукт адсорбируется
хуже, чем др. компоненты газовой смеси, и тогда он содержится в выходящем
газовом потоке. По первому варианту работают, напр., рекуперационные установки
на вискозных произ-вах, улавливающие из отходящих газов и возвращающие
в цикл CS2. Производительность таких установок достигает сотен
тысяч м3 очищаемого газа в час; адсорбент-активный уголь с не
слишком тонкими микропорами, т.е. уголь, в к-ром константа E по
ТОЗМ (см. выше) равна 20-25 кДж/моль. Это значение E0 соответствует
не слишком крутой изотерме, что обеспечивает хорошие условия регенерации.
Такие угли наз. рекуперационными. Десорбцию осуществляют острым водяным
паром. Для экономии энергии холодные и газовые горячие потоки пропускают
через теплообменники.
Очень важна осушка газов и жидкостей, напр. нефтяных газов перед их
переработкой или прир. газов перед транспортировкой; адсорбенты-силикагель
или цеолиты. Десорбцию осуществляют нагреванием. Т. к. десорбция цеолита
связана с большими энергозатратами, применяют комбинированный адсорбент:
осн. массу влаги поглощают легко регенерируемым силикагелем, а глубокую
доосушку-цеолитом.
При тепловой регенерации полный цикл включает А., нагрев адсорбента,
его десорбцию и охлаждение. Большое число стадий обусловливает низкую интенсивность
и высокую энергоемкость процесса. Поэтому широкое распространение получили
т. наз. короткоцикловые установки, весь цикл в к-рых занимает неск. минут.
В них газ подается в адсорбер под значит. давлением, к-рое затем сбрасывается,
и происходит десорбция. Весь процесс идет почти изотермически (отклонение
от изотермичности вызывается только выделением теплоты А. и поглощением
теплоты при десорбции). Стадии цикла: А., сброс давления, десорбция, подъем
давления. Пример-установки с цеолитом для получения воздуха, обогащенного
кислородом.
В установках сдвижущимся слоем адсорбента (в т. наз. гиперсорберах)
последний под действием силы тяжести медленно опускается, выводится из
ниж. части адсорбера и попадает в т. наз. эрлифт, представляющий собой
вертикальную трубу, параллельную адсорбц. колонне. По этой трубе снизу
вверх движется поток воздуха, к-рый поднимает зерна адсорбента в верх.
часть колонны. Перерабатываемый газовый поток поступает в среднюю часть
адсорбера и движется вверх противотоком к адсорбенту. В верхней части колонны
непрерывно происходит А., в нижней - регенерация адсорбента (см. также
Адсорбционная
очистка).
В установках с псевдоожиженным ("кипящим") слоем адсорбента газовый
поток, поступающий в адсорбер снизу, приводит адсорбент во взвешенное состояние.
При этом резко увеличивается эффективность массообмена между адсорбентом
и газом и сокращается длительность А. и десорбции. Такие установки имеют
высокую производительность. Их широкому распространению препятствуют высокие
требования, предъявляемые к мех. прочности зерен адсорбента (недостаточная
прочность обусловливает значит. потери адсорбента вследствие его истирания
и уноса из аппарата).
Осн. требования к адсорбентам: большая адсорбц. емкость, т.е. они должны
представлять собой дисперсные тела с большой уд. пов-стью или с большим
объемом пор; хим. природа пов-сти должна обеспечивать эффективную А. данных
в-в в данных условиях; хим. и термич. стойкость, регенерируемость, доступность.
наиб. распространение получили активные угли, ксерогели нек-рых оксидов
(силика-гели, алюмогели и др.), цеолиты; из непористых адсорбентов-техн.
углерод (сажа) и высокодисперсный SiO2 (аэросил, "белая сажа").
Области применения адсорбционной техники
На явлении А. основаны мн. способы очистки воздуха от вредных примесей
(см. Газов очистка), воды (см. Водоподготовка), а также сахарных
сиропов при сахароварении, фруктовых соков и др. жидкостей в пищ. пром-сти,
отработанных смазочных масел. Удаление влаги как вредной примеси из газов
и жидкостей с помощью твердых адсорбентов-одна из важных отраслей адсорбц.
техники (см. также Газов осушка).
На адсорбц. процессах основано тонкое разделение смесей в-в и выделение
из сложных смесей определенных компонентов. Примеры-разделение изомеров
алканов с целью получения нормальных углеводородов для произ-ва ПАВ, разделение
нефтей при произ-ве моторных топлив. Для газовых смесей адсорбц. методы
разделения используют при получении воздуха, обогащенного кислородом (вплоть
до почти чистого О2); во мн. случаях эти методы успешно конкурируют
с ректификационным (см. Воздуха разделение).
Быстро развивающаяся область применения адсорбц. техники-медицина, где
она служит для извлечения вредных в-в из крови (метод гемосорбции) и др.
физиол. жидкостей. Высокие требования к стерильности ставят очень трудную
задачу подбора подходящих адсорбентов. К ним относятся специально приготовленные
активные угли.
===
Исп. литература для статьи «АДСОРБЦИЯ»: Брунауэр С., Адсорбция газов и паров, пер. с англ., т.
1, М., 1948; де Бур Я, Динамический характер адсорбции, пер. с англ., М.,
1962; Адсорбция и пористость, под ред. М. М. Дубинина [и др.], М., 1976;
Кельиев Н. В., Основы адсорбционной техники, 2 изд., М., 1984; Young D.M.,
Crowell A.D., Physical adsorption of gases, L., 1962. М.М.Дубинин, В.В.
Серпинский.
Страница «АДСОРБЦИЯ» подготовлена по материалам химической энциклопедии.
|



