|
ФУНКЦИИ КИСЛОТНОСТИ,
определяют протонирую-щую способность р-ров к-т. Если протонируется нейтральная
молекула В, Ф. к. обозначают H0, если анион или катион - H_
или H+ соотв. Ф. к. характеризуют равновесие протонирова-ния
в зависимости от концентрации к-ты и позволяют рассчитывать степени протонирования
реагентов в хим. р-ции, ионизирующихся по тому же механизму, что и применяемые
для измерений Ф. к. стабильные соед. (индикаторы).
Ф.к. H0 отвечает
условно записанному равновесию BH+  В + H+, где BH+ - протонир. форма основания В. Константа
равновесия этого процесса
В + H+, где BH+ - протонир. форма основания В. Константа
равновесия этого процесса
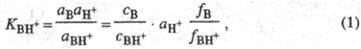
где аB,
aH+ и аBH+ - термодинамич. активности соотв.
основания В, протона H+ и протонир. формы BH+; с -
их равновесные
концентрации; f- коэф. активности. 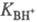 наз. константой основности соед. В.
наз. константой основности соед. В.
Соотношение 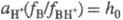 наз. кислотностью среды. Ф-ция кислотности H0 = -lg h0
- При логарифмировании выражения (1) получим соотношение
наз. кислотностью среды. Ф-ция кислотности H0 = -lg h0
- При логарифмировании выражения (1) получим соотношение 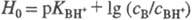 ,
где ,
где 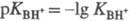 . При
измерении Ф. к. исходят из предположения, что в концентрир. водных р-рах сильных
к-т изменение отношения . При
измерении Ф. к. исходят из предположения, что в концентрир. водных р-рах сильных
к-т изменение отношения 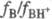 не зависит от хим. природы основания-индикатора В (постулат Гаммета).
не зависит от хим. природы основания-индикатора В (постулат Гаммета).
Аналогично измеряют Н_
и H+,.исходя из след, ур-ний:
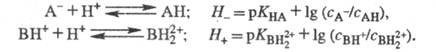
Для определения Ф. к. обычно
используют индикаторный метод, при этом необходимо знать константу основности
индикатора и отношение концентраций его непротонир. и протонир. форм (измеряется
спектрофотометрически или др. методом). Посредством одного индикатора можно
измерить H0 в пределах 2-3 единиц ее изменения, в более
широких диапазонах кислотностей применяют серии индикаторов. Для установления
шкалы кислотности используют в осн. производные нитроанилина.
За стандартное состояние
для шкалы кислотности берут бесконечно разб. водный р-р сильной к-ты, где коэф.
активности fB и fBH+ принимаются равными
единице, а активность протонов равной их концентрации. В таких р-рах H0
численно равна рН.
Индикаторный метод измерения
H0 состоит в следующем. Сначала выбирают наиб. сильное
основание В, к-рое практически полностью протонируется в очень разб. р-рах к-ты
НА. Измерив отношение сB/сBH+, из зависимости lg (сB/сBH+)
от моляльной концентрации МНА к-ты определяют 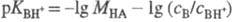 . В таких р-рах -lg МНА = рН, а соблюдение этой зависимости
подтверждает, что в данном интервале концентраций к-ты коэф. активности не изменяются.
Для определения Ф.к. более концентрир. р-ров выбирают след. индикатор таким
образом, чтобы, являясь более слабым основанием, он начинал протонироваться
в тех р-рах к-ты, где первый индикатор еще не полностью протонирован. В этом
"интервале перекрывания" определяют константу основности второго
индикатора D: рКBH+ - рКDH+ = = lg
(cD/cDH+) - Ig (сB/сBH+). Далее,
измеряя cD/cDH+ в более концентрир. р-рах к-ты и зная
pKDH+, определяют H0 для
этих р-ров. Затем аналогичную процедуру проводят со след. индикатором и т. д.
Таким образом, для измерения H0 необходима серия индикаторов.
Так, для системы HCl - H2O (до 16 M HCl) нужно 7-8 индикаторов, для
водных р-ров H2SO4 от разбавл. до 100% H2SO4
- 10-12 индикаторов.
. В таких р-рах -lg МНА = рН, а соблюдение этой зависимости
подтверждает, что в данном интервале концентраций к-ты коэф. активности не изменяются.
Для определения Ф.к. более концентрир. р-ров выбирают след. индикатор таким
образом, чтобы, являясь более слабым основанием, он начинал протонироваться
в тех р-рах к-ты, где первый индикатор еще не полностью протонирован. В этом
"интервале перекрывания" определяют константу основности второго
индикатора D: рКBH+ - рКDH+ = = lg
(cD/cDH+) - Ig (сB/сBH+). Далее,
измеряя cD/cDH+ в более концентрир. р-рах к-ты и зная
pKDH+, определяют H0 для
этих р-ров. Затем аналогичную процедуру проводят со след. индикатором и т. д.
Таким образом, для измерения H0 необходима серия индикаторов.
Так, для системы HCl - H2O (до 16 M HCl) нужно 7-8 индикаторов, для
водных р-ров H2SO4 от разбавл. до 100% H2SO4
- 10-12 индикаторов.
Численные значения Ф.к.
связаны с силой к-ты. Так, 100%-ная H2SO4 имеет H0
= -11,94, чистая CH3SO3H-H0 = -7,86.
Ф. к., судя по нек-рым
данным, мало зависят от т-ры. Однако при повышении т-ры могут изменяться 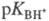 оснований.
оснований.
Ф. к. описывают только
процесс протонирования. Во мн. случаях, кроме этого процесса, ионизация основания
может заключаться также в образовании ионных пар, разл. комплексов протонир.
формы с молекулами р-рителя, димеров и т.д. В таких случаях для корректного
использования Ф.к. при описании ионизации слабого основания необходимо знать
механизм его ионизации и измерять равновесные концентрации всех образующихся
форм основания В. На практике обычно измеряют равновесную концентрацию только
одной формы (обычно - неионизированной), а концентрацию формы BH+
вычисляют из ур-ния баланса концентраций, тем самым постулируя простой механизм
протонирования. Такая практика, а также недоказанность постулата Гаммета привели
к появлению ряда Ф.к., относящихся к разл. классам хим. соед. (HА
- Ф. к. амидов, 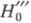 -
третичных аминов, HI -индолов, НB - бензофенонов,
НM - азуленов и т.д.). Нек-рые из
этих Ф. к. связаны с иным, чем протонирование, механизмом ионизации. Это м.
б. образование ионных пар типа BH+·A- (А- -
анион к-ты), к-рое имеет место наряду с протонированием ( -
третичных аминов, HI -индолов, НB - бензофенонов,
НM - азуленов и т.д.). Нек-рые из
этих Ф. к. связаны с иным, чем протонирование, механизмом ионизации. Это м.
б. образование ионных пар типа BH+·A- (А- -
анион к-ты), к-рое имеет место наряду с протонированием (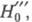 HI), либо образование комплекса B·HA (НM).
Возможны случаи, когда действительно не соблюдается постулат Гаммета.
HI), либо образование комплекса B·HA (НM).
Возможны случаи, когда действительно не соблюдается постулат Гаммета.
Развивая этот подход, P.
Кокс и К. Эйтс (1978) ввели понятие "избыточная кислотность" X,
к-рая отражает разность между наблюдаемой (формально измеренной) Ф. к. для данного
класса соед. и "идеальной" Ф.к., строго подчиняющейся постулату
Гаммета (H0 , Н_). Отклонения от соблюдения постулата
Гаммета выражаются ур-нием:
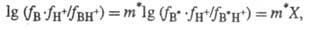
где В* - "идеальное"
основание; В - основание из данного ряда; m* - параметр, характерный
для данного класса соед. Практическое применение избыточной кислотности X
основано на линейном соотношении:
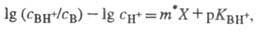
где CH+
- равновесная концентрация протонов в р-ре.
Схема протонирования слабого
основания (В + H+ BH+)
не есть реальное хим.
равновесие в р-рах к-т. Протон в таких р-рах существует в виде простейшего устойчивого
гидрата BH+)
не есть реальное хим.
равновесие в р-рах к-т. Протон в таких р-рах существует в виде простейшего устойчивого
гидрата 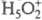 , а протонирование основания В есть р-ция замещения: В + H5O2+
, а протонирование основания В есть р-ция замещения: В + H5O2+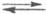 BHOH2+
+ H2O. Индикаторные отношения BHOH2+
+ H2O. Индикаторные отношения 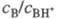 и константы основности вычисляют так же, как и при формальной схеме протонирования:
и константы основности вычисляют так же, как и при формальной схеме протонирования:
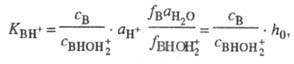
где 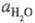 - активность воды. Присутствие
- активность воды. Присутствие 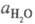 в ф-ле для кислотности
объясняет известный факт независимости отношения
в ф-ле для кислотности
объясняет известный факт независимости отношения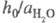 от природы сильной к-ты (для других Ф. к. это не
соблюдается).
от природы сильной к-ты (для других Ф. к. это не
соблюдается).
Дигидрат протона 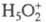 и протонир. форма.
и протонир. форма.  образованы
сильной симметричной водородной связью центр, протона: (Н2О...Н...ОН2)+
и (В...Н...ОН2)+. Геом. и энергетич. параметры этой связи
не зависят от структуры участвующих в ней частиц, а ее энергия разрыва достаточно
велика (134,1-142,5 кДж/моль). Этим можно объяснить соблюдение постулата Гаммета.
Отклонения от соблюдения постулата, в частности, м. б. объяснено образованием
другой протонир. формы по схеме: В + H5O2 образованы
сильной симметричной водородной связью центр, протона: (Н2О...Н...ОН2)+
и (В...Н...ОН2)+. Геом. и энергетич. параметры этой связи
не зависят от структуры участвующих в ней частиц, а ее энергия разрыва достаточно
велика (134,1-142,5 кДж/моль). Этим можно объяснить соблюдение постулата Гаммета.
Отклонения от соблюдения постулата, в частности, м. б. объяснено образованием
другой протонир. формы по схеме: В + H5O2 BH+
+ 2H2O. Такие случаи имеют место, если разность сродства к протону
молекул В и H2O превышает 113 кДж/моль. BH+
+ 2H2O. Такие случаи имеют место, если разность сродства к протону
молекул В и H2O превышает 113 кДж/моль.
Корректное использование
Ф.к. для расчетов степеней протонирования и констант основности хим. соед. в
р-рах к-т должно быть основано, во-первых, на установлении механизма ионизации
данного соед., во-вторых, на уверенности в том, что образующаяся протонир. форма
есть ион, образованный сильной симметричной водородной связью.
Понятие Ф.к. введено Л.
Гамметом в 1932.
===
Исп. литература для статьи «ФУНКЦИИ КИСЛОТНОСТИ»: Гаммет Л.
П., Основы физической органической химии, пер. с англ., M., 1972; Либрович H.Б.,
"Изв. АН СССР. Сер. хим.", 1990, № 1, с. 32-35; Rochester C.H.,
Acidity functions, L.-N. Y., 1970; Cox R. А., Yates K., "J. Amer.
Chem. Soc.", 1978, v. 100, № 12, p. 3861-67.
H. Б. Либрович.
Страница «ФУНКЦИИ КИСЛОТНОСТИ» подготовлена по материалам химической энциклопедии.
|



