|
АЗОСОЕДИНЕНИЯ, содержат азогруппу — N=N—, связанную с атомами
С двух одинаковых или разных орг. остатков. Общая ф-ла RN=NR'.
Названия А., у к-рых R=R', образуют, ставя частицу "азо" перед названием
соединения, остатки к-рого входят в молекулу А., напр. азометан (R = R'
= СН3), 1,1-азонафталин (R = R'= 1-нафтил). Если R и R'-разные,
частицу "азо" располагают в середине названия А., напр. нафталин-2-азобензол
(R = 2-нафтил, R' = C6H5), бензолазоацетоацетани-лид
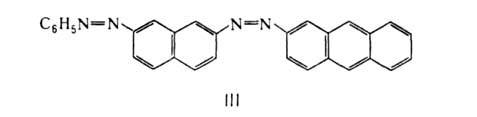
(ф-ла I), З-метил-1-фенил-5-пиразолон-4-азобензол (II).
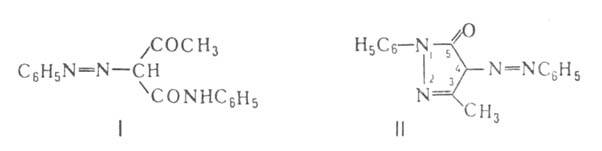
Группу ArN=N— можно рассматривать как заместитель. Тогда названия соответствующих
А. образуются, напр., так: n-фенилазобензолсульфокислота n-(C6H5N=N)—
—C6H4SO3H, 2-(7-фенилазо-2-нафтилазо)антрацен
(III):
Атомы N в азогруппе находятся в состоянии sp2-гибридизации,
причем две из трех 5р2-орбиталей каждого атома участвуют в образовании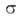 -связей,
на третьей располагается неподеленная пара электронов, а -связей,
на третьей располагается неподеленная пара электронов, а -связь
образуется за счет pz--орбиталей. В результате азогруппа нелинейна,
что обусловливает существование цис- и транс-изомеров А.
Стабильна транс-форма, к-рая превращается в цис-изомер при
облучении р-ров светом длиной волны, соответствующей полосе поглощения
облучаемого А. -связь
образуется за счет pz--орбиталей. В результате азогруппа нелинейна,
что обусловливает существование цис- и транс-изомеров А.
Стабильна транс-форма, к-рая превращается в цис-изомер при
облучении р-ров светом длиной волны, соответствующей полосе поглощения
облучаемого А.
Наличие неподеленных электронов создает возможность электронного п
->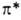 -перехода
в азогруппе, приводящего к появлению полосы поглощения в электронном спектре:
у алифатич. азосоединений - в УФ-области (160-ЗЮнм), у ароматических в
результате сопряжения с кольцами-в длинноволновой области, напр. при 432
и 450 нм у цис-и транс-азобензолов соотв.; эти полосы характеризуются
низкой интенсивностью, т. к. п -> -перехода
в азогруппе, приводящего к появлению полосы поглощения в электронном спектре:
у алифатич. азосоединений - в УФ-области (160-ЗЮнм), у ароматических в
результате сопряжения с кольцами-в длинноволновой области, напр. при 432
и 450 нм у цис-и транс-азобензолов соотв.; эти полосы характеризуются
низкой интенсивностью, т. к. п ->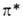 -переход
в азогруппе запрещен по симметрии. Азогруппа обусловливает также появление
интенсивной полосы -переход
в азогруппе запрещен по симметрии. Азогруппа обусловливает также появление
интенсивной полосы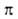 -> ->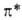 -перехода
у ароматич. А. в области 280-320 нм. Введение электронодонорного заместителя
в сопряженное с азогруппой положение смещает полосу -перехода
у ароматич. А. в области 280-320 нм. Введение электронодонорного заместителя
в сопряженное с азогруппой положение смещает полосу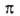 -> ->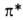 -перехода
в видимую область спектра и А. становится типичным красителем; введение
электроноак-цепторного заместителя в сопряженное с азогруппой положение
второго ароматич. остатка еще более усиливает это смещение. Соответствующим
подбором заместителей в разных ароматич. остатках А. можно добиться значит.
углубления цвета А. (см. Цветность органических соединений). Этот
прием используется в синтезе азокрасителей. В ИК-спектрах характеристич.
полосы поглощения vN=N у цис-изомеров и несимметричных
А. лежат в области 1400-1600 см -1; у ароматич. А. они перекрываются
полосами поглощения колец. -перехода
в видимую область спектра и А. становится типичным красителем; введение
электроноак-цепторного заместителя в сопряженное с азогруппой положение
второго ароматич. остатка еще более усиливает это смещение. Соответствующим
подбором заместителей в разных ароматич. остатках А. можно добиться значит.
углубления цвета А. (см. Цветность органических соединений). Этот
прием используется в синтезе азокрасителей. В ИК-спектрах характеристич.
полосы поглощения vN=N у цис-изомеров и несимметричных
А. лежат в области 1400-1600 см -1; у ароматич. А. они перекрываются
полосами поглощения колец.
Алифатич. А. при нагр. распадаются с выделением N2 и образованием
своб. радикалов, благодаря чему, напр., 2,2'-азо-бис-изобутиронитрил,
гладко
разлагающийся при 60-100°С или облучении УФ-светом, применяется как инициатор
свободнорадикальных р-ций. Простейший представитель алифатич. А.-азометан
- при осторожном нагревании устойчив до 400 °С, но при быстром нагревании
разлагается со взрывом. Получают алифатич. А. гл. обр. дегидрированием
диалкилгидразинов действием окислителей типа К2Сr2О7
или HgO, напр.: CH3NH—NHCH3 + + [О] -> CH3N=NCH3
+ Н2О.
А., содержащие алифатич. иаромати ч. остатки, более устойчивы вследствие
сопряжения азогруппы с ароматич. ядром, напр. бензолазоэтан C6H5N=NC2H5-жидкость,
кипящая при 180°С со слабым разложением. Получают их аналогично алифатич.
А. из N-арил-N'-алкилгидразинов, напр.: C6H5NH—NHCH3
+ [О] -> C6H5N=NCH3 + + Н2О.
Ароматич. А., а также гетероароматич. и смешанные, в к-рых азогруппа
сопряжена с обоими остатками, как в IV и V (таутомерные енольные формы
соединений I и II соотв.), устойчивы.
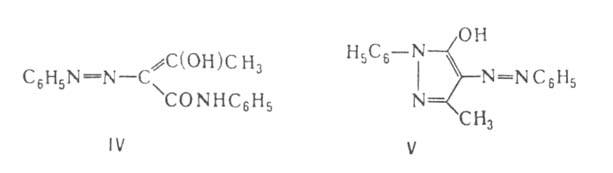
Простейший представитель ароматам. А.-азобензол С6Н5М:=:МС6Н5
-
мол. м. 182,22; оранжево-красные кристаллы; т. пл. 68°С, т. кип. 293°С;
не раств. в воде, раств. в этаноле, эфире, бензоле, хлороформе.
При наличии в молекуле А. сопряженного с азогруппой заместителя, содержащего
подвижный атом Н (напр., ОН, NH2), возможна таутомерия азоидной
и хинонгидразонной форм (азо-гидразонная таутомерия), напр.:
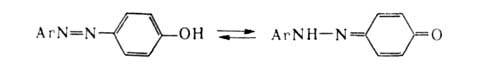
Положение равновесия зависит от хим. строения А.; так, объемистые заместители
в орто- и пара-положениях к азогруппе затрудняют переход
в хинонгидразонную форму, поскольку нарушают планарность молекулы, создавая
пространств. затруднения. Большое значение имеют также полярность р-рителя
и рН среды; напр., 1-гидроксинафта-лин-4-азобензол в пиридине и в водно-щелочной
среде существует почти исключительно в азоидной форме, в нитробензоле и
уксусной к-те - преим. в хинонгидразонной, а в бензоле-в виде смеси приблизительно
одинаковых кол-в обеих форм.
Мягкие восстановители (Na2S, соли Fe2+ в водном
NH3 и др.) в мягких условиях не восстанавливают азогруппу; это
позволяет, напр., не затрагивая последнюю, восстановить нитрогруппу до
NH2. Более сильные восстановители и в более жестких условиях
(напр., Na2Sx ; Sn, Zn, Fe, SnCl2 или
ТiС13 с соляной к-той; VSO4 с H2SO4;
Na2S2O4 в щелочной среде; Н2
над катализатором) расщепляют азогруппу, напр.:

Эта р-ция используется в произ-ве нек-рых аминов, для установления строения
А. и их количеств. анализа, для вытравной печати узоров на тканях, окрашенных
азокрасите-лями. Р-ция в нек-рых случаях м. б. остановлена на стадии образования
гидразосоединений.
При действии сильных минер. к-т азогруппа способна присоединять протон
и превращаться в гидразонную группу с переносом положит. заряда на остаток,
связанный с азогруппой, напр.:
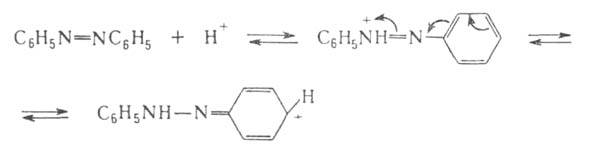
Т. к. основные св-ва азогруппы очень слабы, протонирован-ное производное
уже под действием воды распадается с образованием исходного соединения.
Мягкие окислители, напр. Н2О2 в уксуснокислом
р-ре, окисляют ароматич. А. до азоксисоединений, сильные (дымящая
HNO3 на холоду или С12)- расщепляют А. по р-циям:
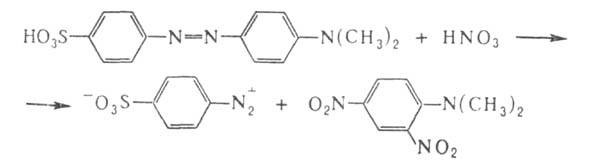 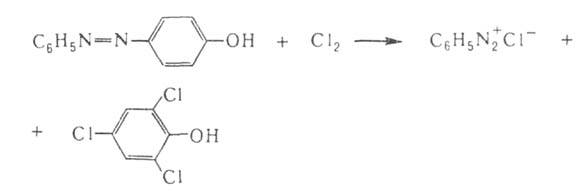
Это используется для установления строения А. Аналогично, но медленнее
действует HNO2, взятая в избытке. Длительное кипячение с водой
постепенно разрушает мн. ароматич. А.; процесс ускоряется при нагр. с разб.
к-тами и щелочами. Конц. H2SO4 при умеренных т-рах
сульфирует нек-рые А., но при повыш. т-рах полностью разрушает их.
Осн. способ получения ароматич. А.-азосочетание, в т.ч. окислительное.
Практич. значение имеют также: взаимод. первичных аминов с нитрозосоединениями
(ArNO + + Ar'NH2 -> ArN=NAr' + Н2О) или нитросоединениями
с послед. восстановлением образовавшихся азоксисоединений
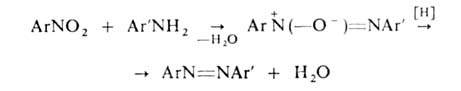
восстановление нитро-, нитрозо- и азоксисоединений, напр. ArNO2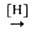 ArN=NAr
+ H2O; ArNO ArN=NAr
+ H2O; ArNO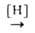 ArN=NAr
+ Н2О; окисление первичных аминов: ArN=NAr
+ Н2О; окисление первичных аминов: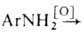 -> ArN=NAr + Н2О.
-> ArN=NAr + Н2О.
В лаб. А. могут быть также получены взаимод. арилгидразинов с хинонами,
окислением нек-рых диазосоед. и др. Ароматич. А. широко применяют как азокрасители.
Количеств, анализ А. основан на восстановит. расщеплении азогруппы при
титровании р-рами VSO4, TiCl3, SnCl2 и
др.; применяются также колориметрич., спектрофотометрич. и др. оптич. методы.
Качеств. контроль осуществляется тонкослойной и бумажной хроматографией.
А. открыты в 1834 Э. Мичерлихом, получившим азобензол восстановлением
C6H5NO2 в щелочной среде.
===
Исп. литература для статьи «АЗОСОЕДИНЕНИЯ»: Вснкатарама" К., Химия синтетических красителей, пер. с
англ., т. 1, Л., 1956; Химия синтетических красителей, под ред. К. Венкатарамана,
пер. с англ., т. 3-6, Л., 1974-77; Цоллингер Г., Химия азокрасителей, пер.
с нем., Л., 1960; Порай-Кошиц Б. А., Азокрасители, Л., 1972; Степанов Б.И.,
Введение в химию и технологию органических красителей, 2 изд., М., 1977;
Аналитическая химия синтетических красителей, под ред. К. Венкатарамана,
пер. с англ., Л., 1979. Б. И. Степанов.
Страница «АЗОСОЕДИНЕНИЯ» подготовлена по материалам химической энциклопедии.
|



