ПРОГРЕСС В ОБЛАСТИ ОКИСЛИТЕЛЬНОГО КАТАЛИЗА (I часть)
Представляем технологию окисления бензола в фенол закисью азота, разрабатываемую в новосибирском Институте катализа им. Г. К. Борескова СО РАН.
 Кислород является самым распространенным химическим элементом на Земле. Если учитывать достаточно полно исследованную часть планеты, то из более чем 100 известных элементов свыше половины ее состава - 53,6 % (ат.) занимает кислород [1]. Поэтому неудивительно, что реакции с участием О2 играют столь большую роль в окружающей нас среде, и особенно в живой природе. Биологический цикл кислорода можно свести к его присоединению в животном мире (процессдыхания) и выделению в растительном мире (процесс фотосинтеза). Поддержание этого тонкого баланса, на острие которого и существует жизнь, достигается через множество окислительно-восстановительных реакций, регулируемых ферментами уникальными катализаторами, над совершенством которых Природа трудилась миллионы лет.
Кислород является самым распространенным химическим элементом на Земле. Если учитывать достаточно полно исследованную часть планеты, то из более чем 100 известных элементов свыше половины ее состава - 53,6 % (ат.) занимает кислород [1]. Поэтому неудивительно, что реакции с участием О2 играют столь большую роль в окружающей нас среде, и особенно в живой природе. Биологический цикл кислорода можно свести к его присоединению в животном мире (процессдыхания) и выделению в растительном мире (процесс фотосинтеза). Поддержание этого тонкого баланса, на острие которого и существует жизнь, достигается через множество окислительно-восстановительных реакций, регулируемых ферментами уникальными катализаторами, над совершенством которых Природа трудилась миллионы лет.
Хотя искусственные катализаторы далеко не так совершенны, как ферменты, окислительные реакции играют большую роль и в нашей технической деятельности. В течение четырех последних десятилетий в области окислительного катализа достигнуты огромные успехи. Они привели к созданию почти двух десятков новых технологий [2—5], среди которых такие широко известные процессы, как окисление пропилена в акролеин и акриловую кислоту, эпоксидирование пропилена, окисление бутилена и бутана в малеиновый ангидрид и др. Эти процессы в значительной мере изменили саму структуру химической промышленности. Отметим, что один из этих процессов – окислительное ацилирование этилена в винилацетат – создан на базе открытия, сделанного в России. Реакция, лежащая в его основе, была впервые проведена в 1960 г. И.И. Моисеевым и соавт.
Особо следует выделить роль окислительного катализа как главного инструмента нейтрализации вредных химических отходов с целью защиты окружающей среды. Успехи экологического катализа в значительной мере помогли изменить образ химии в глазах общества от губителя природы до ее друга и спасителя.
Развитие работ как в области селективного окисления, так и в области защиты окружающей среды рассмотрено в целом ряде книг и обзоров, с которыми периодически выступают ведущие специалисты по катализу [7—15]. Но есть область окислительного катализа, в которой, несмотря на большие усилия, успехи остаются весьма скромными. Это реакции окислительного гидроксилирования парафинов и ароматических соединений. Потребность в таких реакциях велика - это cинтез разнообразных спиртов и фенолов. В настоящее время их получают путем сложных многостадийных процессов. В качестве примера можно привести две простейшие реакции этого типа, которые нередко включают в число десяти труднейших проблем современной химии [16]: окисление метана в метанол и окисление бензола в фенол.
Для проведения первой реакции метан сначала превращают в СО и Н2, а окисление бензола в фенол осуществляют через трехстадийный кумольный процесс.В то же время в живой природе эти реакции легко протекают с участием ферментов монооксигеназ в однустадию путем простого присоединения одного атома кислорода к окисляемой молекуле [17]. Попытки понять и воспроизвести уникальную способность монооксигеназ наталкиваются на трудность, связанную с созданием искусственных систем, которые обеспечивали бы столь эффективную активацию кислорода [18, 19].
Для достижения лучших результатов исследователи пытаются вместо О2 применить нетрадиционные окислители в виде различных кислородсодержащих молекул. Среди них наибольшее внимание привлекают пероксид водорода Н2О2 и закись азота N2O, с использованием которых связаны недавние успехи в этой трудной области. Наибольший прогресс достигнут в отношении реакции окисления бензола в фенол.
ПРОМЫШЛЕННЫЕ СПОСОБЫ ПОЛУЧЕНИЯ ФЕНОЛА
Краткая историческая справка:
История фенола насчитывает уже более 160 лет. Впервые он был выделен из каменноугольной смолы в 1834 г., откуда и получил свое название –карболовая (угольная) кислота. Быстрый рост потребления фенолапоставил вопрос об искусственных способах его получения, создание которых является одной из самых ярких страниц в истории органической химии. Ниже кратко описаны промышленные процессы, которые в то или иное время использовались для получения фенола.
1. Сульфонатный процесс был первым фенольным процессом, реализованным в промышленном масштабе фирмой «BASF» в 1899 г. [20]. Этот метод основан на сульфировании бензола серной кислотой с последующим щелочным плавлением сульфокислоты:
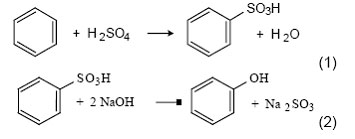
Несмотря на применение агрессивных реагентов и образование большого количества отходов сульфита натрия, данный метод использовался в течение почти 80 лет. В США это производство было закрыто лишь в 1978 году.
2. В 1924 г. фирмой «Dow Chemical» (США) был разработан процесс получения фенола, включающий реакцию хлорирования бензола и последующий гидролиз монохлорбензола:
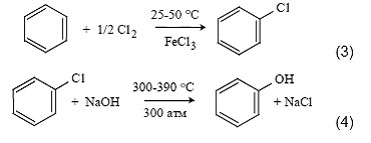
Независимо аналогичная технология была разработана немецкой фирмой «I.G. Farbenindustrie Co». Какотмечает Kirk Othmer [21], захват этой технологии составлял цель нескольких научных разведывательных групп стран-победительниц, прибывших в Германию сразу после окончания Второй мировой войны. Отметим, что в составе одной из таких групп был Г.К. Боресков.
Впоследствии стадия получения монохлорбензола и стадия его гидролиза были усовершенствованы, и процесс получил название «процесс Рашига». Суммарный
выход фенола по двум стадиям составляет 70—85%.
3. Циклогексановый процесс, разработанный фирмой «Scientific Design Co.», основан на окислении циклогексана в смесь циклогексанона и циклогексанола которая далее дегидрируется с образованием фенола
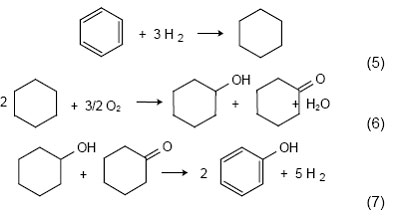
В 60-е годы фирма «Monsanto» в течение нескольких лет использовала этот метод на одном из своих заводов в Австралии, однако в дальнейшем перевела его накумольный способ получения фенола.
4. В 1961 г. фирмой «Dow Chemical of Canada» был реализован процесс через разложение бензойной кислоты- это единственный способ синтеза фенола, основанный на использовании небензольного сырья:
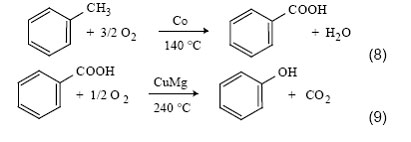
Обе реакции протекают в жидкой фазе. Первая реакция окисление толуола - использовалась в Германии уже в период Второй мировой войны для получения бензойной кислоты. Реакция протекает в довольно мягких условиях с высоким выходом. Вторая стадия является более трудной вследствие дезактивации катализатора и низкой селективности по фенолу. Полагают, что проведение этой стадии в газовой фазе может сделать процесс более эффективным [23]. В настоящее время этот метод используется на практике, хотя его доля в мировом производстве фенола составляет лишь около 5%. Остальные 95% приходятся на кумольный процесс [24].
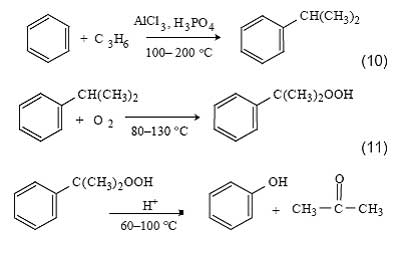
5. Кумольный процесс получения фенола включает три стадии: алкилирование бензола в кумол, окисление кумола в гидропероксид кумола и разложение гидропероксида на фенол и ацетон
Впервые этот способ был разработан в Советском Союзе. Химический маршрут процесса был открыт в 1942 г. группой талантливых химиков, в которую входили П.Г. Сергеев, Р.Ю. Удрис и Б.Д. Кружалов. В это время они были репрессированы и работали в специальной весьма хорошо оборудованной лаборатории, которая одновременно служила и местом заключения. В 1949 году в г. Дзержинске Горьковской области был введен в действие первый в мире кумольный завод. В 1951 г. в связи с успешным пуском завода большой группе советских ученых и инженеров была присвоена высшая награда страны -Сталинская премия. Драматическая история создания кумольного процесса вСССР описана в ряде статей, опубликованных в 80-е годы [28].
В 1947 г. Сергееву, Удрису и Кружалову были выданы авторские свидетельства СССР [26]. К сожалению, их открытие не получило мировой известности. На Западе кумольный метод был разработан в конце 40-х годов и отчасти известен как процесс Хока [27] поимени немецкого ученого, позднее независимо открывшего кумольный путь синтеза фенола. В промышленном масштабе этот метод стал впервые использоваться в США в начале 50-х годов [21]. С этого времени на многие десятилетия кумольный процесс становится образцом химических технологий во всем мире.
В настоящее время производство фенола достигло около 6 млн. тонн в год и продолжает быстро расти. Как таковой, фенол практически не используется. Но благодаря тому, что его молекула включает два умеренно реакционноспособных фрагмента (ароматическое кольцо и ОН-группа), каждый из которых может быть вовлечен в дальнейшие превращения [28], фенол является основой для синтеза многих важных химических продуктов. В табл. 1 на примере США показана структура потребления фенола.

Недостатки кумольного метода
Несмотря на прекрасно отлаженную технологию и длительный опыт эксплуатации, кумольный метод имеет ряд недостатков.
Прежде всего это наличие взрывоопасного промежуточного соединения (гидропероксид кумола), а также многостадийность метода, что требует повышенных капитальных затрат и делает труднодостижимым высокий выход фенола в расчете на исходный бензол. Так, при выходе полезного продукта 95% на каждой из трех стадий итоговый выход составит лишь 86%. Приблизительно такой выход фенола и дает кумольный метод в настоящее время .
Но самый важный и принципиально неустранимый недостаток кумольного метода связан с тем, что в качестве побочного продукта образуется ацетон. Это обстоятельство, которое первоначально рассматривалось как сильная сторона метода, становится все более серьезной проблемой, поскольку ацетон не находит эквивалентного рынка сбыта. В 90-х годах эта проблема стала особенно ощутимой после создания новых способов синтеза метилметакрилата путем окисления углеводородов С4, что резко сократило потребность в ацетоне.
Об остроте ситуации говорит тот факт, что в Японии разработана технология, предусматривающая рецикл ацетона. С этой целью к традиционной кумольной схеме добавляются еще две стадии - гидрирование ацетона в изопропиловый спирт и дегидратация последнего в пропилен [29]:
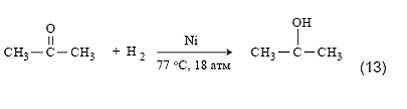
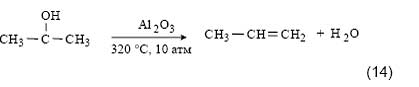
Образующийся пропилен снова возвращают на стадию алкилирования бензола. В 1992 году фирма «Mitsui» пустила крупное производство фенола (200 тыс. т/год), основанное на этой пятистадийной кумольной технологии.
Предлагаются также другие сходные модификации кумольного метода, которые позволили бы смягчить проблему ацетона. Однако все они приводят к значительному усложнению технологии и не могут рассматриваться как перспективное решение проблемы.
Поэтому исследования, ориентированные на поиск новых путей синтеза фенола, которые основывались бы на прямом окислении бензола, в последнее десятилетие приобрели особенно интенсивный характер. Работы ведутся главным образом в следующих направлениях: окисление молекулярным кислородом, окисление моноатомными донорами кислорода и сопряженное окисление.
Отметим, что аналогичные подходы применяются и для поиска эффективных способов селективного окисления низших парафинов, включая метан.
Поиск новых путей синтеза фенола
Окисление молекулярным кислородом. Прямое окисление бензола молекулярным кислородом представляется наиболее привлекательным методом получения фенола:
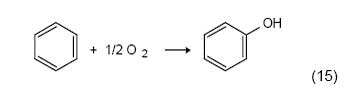
Однако это на первый взгляд самое простое и очевидное решение проблемы оказалось чрезвычайно трудной задачей. Работы по окислению бензола с помощью О2 начались еще до того, как в 1865 г. Кекуле предложил структурную формулу бензольного кольца [30]. С тех пор многочисленные попытки найти эффективный путь для проведения этой реакции не прекращаются. Окисление бензола ведут как в жидкой, так и в газовой фазах, при низком и высоком давлениях, в отсутствие и в присутствии разнообразных катализаторов [31]. Несмотря на отдельные успехи, результаты этих работ пока далеки от практического применения.
Начиная с 80-х годов, значительные усилия предпринимаются для проведения этой реакции в жидкой фазе с использованием в качестве катализаторов различных комплексов переходных металлов, среди которых наибольшую активность проявляют соединения Pd и Cu [32, 33].
Однако после нескольких оборотов реакция, как правило, прекращается вследствие деградации катализатора.
Моноатомные доноры кислорода.
Более успешные результаты дает применение в качестве окислителей так называемых моноатомных доноров кислорода в виде различных кислородсодержащих молекул. Среди таких молекул наибольшее внимание привлекает пероксид водорода:
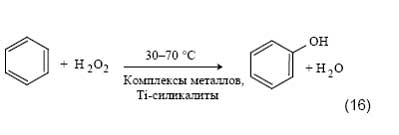
Окисление с помощью Н2О2 проводят в присутствии солей и комплексов переходных металлов, в том числе инкапсулированных в матрице цеолита [34, 35]. Показана возможность проведения этой реакции в условиях межфазного катализа [36].
Исследования в данной области приобрели особенно интенсивный характер после открытия цеолитов состава Ti-Si (TS-1) и их уникальных свойств в реакциях жидкофазного окисления с помощью пероксида водорода [37]. На этой основе фирмой «Enichem» разработан промышленный процесс получения гидрохинона и пирокатехина путем гидроксилирования фенола. Вслед за цеолитами TS-1 были опробованы разнообразные цеолитные системы, содержащие как титан, так и другие переходные металлы [38-40]. Однако, в отличие от окисления фенола, ароматическое кольцо которого активировано присутствием ОН-группы, окисление бензола протекает менее активно и со значительно меньшей селективностью по пероксиду водорода вследствие его побочного разложения на кислород и воду.
Следует отметить, что в любом случае реакция (16) едва ли перспективна для практического использования из-за высокой стоимости Н2О2 (по сравнению со стоимостью фенола).
Помимо пероксида водорода, в исследовательских целях применяется ряд других, более сложных и дорогостоящих монокислорододонорных окислителей, таких как перкислоты, иодозобензол и т.д. Однако для окисления бензола они практически не используются.
Более вероятным окислителем для бензола с практической точки зрения представляется азотная кислота, которая впервые была использована для этой цели в 1925 г. [30]. В более поздних работах было показано, что эффективными катализаторами этой реакции являются оксидные системы на основе V2O5 и MoO3 [41]:
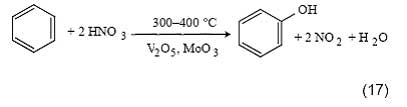
Окисление протекает в газовой фазе в присутствии паров воды при температуре 300-400 оС. Конверсия бензола достигает 52% при селективности по фенолу более 90%. Образующийся диоксид азота может быть окислен до HNO3 и вновь использован для окисления бензола.
Заслуживает упоминания способ окисления бензола, основанный на реакции с сульфатом двухвалентной меди в качестве окислителя:
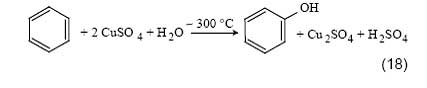
Реакция протекает за счет восстановления Cu2+ в Cu+ или даже в Cu0. Первые патенты в этой области появились в 60-е годы [42]. В последнее время интерес к этой реакции возобновился [43]. Есть сообщение [44] об использовании воды в качестве окислителя бензола:

По данным этой работы, при нормальном давлении и температуре 200-300 оС в присутствии смешанных оксидов Fe, Cu и Mn достигается конверсия бензола 15% с селективностью по фенолу 97%. Однако этот результат требует серьезной проверки.
Вероятнее всего, образование фенола здесь связано не с реакцией (19), протекание которой с таким выходом фенола невозможно в силу термодинамических ограничений, а обусловлено окислением бензола за счет кислорода катализатора. К рассматриваемому классу реакций относится и реакция окисления бензола в фенол с помощью закиси азота, на основе которой создан новый одностадийный способ получения фенола (см. ниже).
Сопряженное окисление
Пероксид водорода может непосредственно образовываться в реакционной системе in situ и тут же расходоваться на окисление субстрата. Этот подход рассматривается как одно из наиболее перспективных направлений не только для окисления ароматических соединений, но и многих других углеводородов, включая парафины [4, 45, 46]. Использование пероксида по мере его образования из Н2 и О2 позволяет значительно увеличить селективность полезного использования H2O2. Это типичный пример сопряженного процесса, протекающего с участием вещества-«жертвы» [47]:
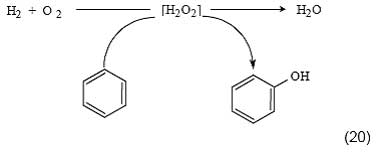
Напомним, что механизм сопряжения заключается в том, что промежуточное вещество, образующееся при протекании первичной реакции, частично используется для осуществления вторичной реакции, которая сама по себе в этих условиях не происходит. В данном случае первичной реакцией является окисление водорода в H2O через промежуточное образование Н2О2, а вторичной реакцией -окисление бензола в фенол.
Работы в этом направлении интенсивно ведутся во многих лабораториях мира с использованием разнообразных катализаторов и приемов [34, 45, 48]. Наиболее эффективные катализаторы включают платину или другой благородный металл, который вместе с оксидом ванадия наносится на силикагель. На таком катализаторе в растворе уксусной кислоты бензол почти со 100%-ной селективностью окисляется в фенол. Однакоселективность реакции по водороду все еще остается невысокой и составляет 10-15% (это значит, что на образование 1 моль фенола расходуется 7-8 моль водорода).
Сопряженное окисление водорода и бензола может протекать также в газовой фазе при повышенной температуре [49]. Помимо окисления водорода, в качестве первичной реакции может выступать процесс окисления других молекул, например монооксида углерода [50]:
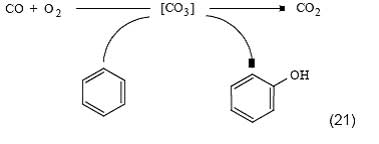
Сопряжение может быть организовано также в сочетании с окислением восстановленной формы металла в растворах:
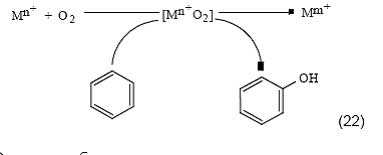
Окисление бензола здесь стимулируется окислительными превращениями металлов: Fe2+ → Fe3+, Sn2+ → Sn4+ [51], Cu+ → Cu2+ [52], Zn0 → Zn2+ [53].
Иногда, как в случае использования цинковой пыли, для протекания реакции требуется введение в систему дополнительного катализатора, в качестве которого могут выступать соли переходных металлов. Недостаток сопряженных реакций состоит в трудности подбора таких условий, которые были бы оптимальными как для образования сопрягающего промежуточного соединения в первичной реакции, так и для его эффективного использования во вторичной реакции. В последние годы широкое развитие получают работы по исследованию реакций сопряженного окисления углеводородов в электрохимических ячейках, представляющих собой реакторы типа топливных элементов [54—56].
Проведение реакций в таких устройствах открывает дополнительную возможность для управления реакцией посредством регулирования электрического тока, пропускаемого через ячейку.В заключение данного раздела отметим, что реакция окисления бензола в фенол, вероятно, является рекордсменом по разнообразию подходов, количеству испытанных каталитических систем, а также по объему усилий, предпринимаемых исследователями для осуществления этой реакции. Сравниться может только реакция окисления метана в метанол, которая является столь же простой на первый взгляд и столь же трудной для реального воплощения, как и окисление бензола.
Осуществление этих реакций, несомненно, останется заветной мечтой и для химиков XXI века.
Открытие цеолитных катализаторов для окисления бензола закисью азота Закись азота была открыта Пристли в конце 18 века. Вслед за Дэви, который в 1799 г. изучил ее физиологические свойства, закись азота называют «веселящим газом», поскольку вдыхание N2O приводит человека в веселое расположение духа. Благодаря легкому наркотическому действию, ее нередко используют в качестве анестезирующего средства.
Термодинамически N2O - неустойчивая эндотермическая молекула, свободная энергия образования которой равна ΔG fo =103,6 кДж/моль. Однако кинетически закись азота весьма стабильна, ее некаталитическое разложение становится заметным лишь при температуре выше 600 оС. В 1983 году Ивамото с сотруд. впервые использовал
закись азота для окисления бензола в фенол [57]:
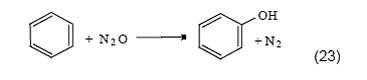
Эта реакция позволила выйти на принципиально новый уровень селективности по фенолу. При температуре 550 оС и добавлении в реакционную смесь 30% H2O селективность по фенолу на нанесенных ванадиевых катализаторах достигала 71% при конверсии бензола 10%.
Этот результат вызвал со стороны ученых оживленные отклики, в которых особо подчеркивалась экологическая безопасность нового процесса и давались весьма оптимистические оценки относительно его практической перспективы [58, 59]. Однако пилотные испытания, проведенные в реакторе с кипящим слоем катализатора, не подтвердили эти ожидания. Селективность ванадиевых катализаторов оказалась недостаточно высокой для практического применения процесса.
Тем не менее, работа Ивамото и сотруд. [57] стимулировала поиск новых более эффективных катализаторов. Испытания огромного числа каталитических систем выявили чрезвычайно высокую требовательность реакции окисления бензола в фенол закисью азота. Катализаторы оказывались либо неактивными, либо неселективными [60].
В 1988 г. три группы исследователей – Сузуки с соавт. [61] (Токийский технологический институт), Губельманн и Тирель [62] (фирма «Rhone Poulenc») и Харитонов с соавт. [63] (Институт катализа СО РАН) независимо показали, что лучшими катализаторами этой реакции являются цеолиты ZSM-5. На цеолитах реакция протекает при значительно более низкой температуре и, что особенно важно, с селективностью по фенолу, близкой к 100%. Несмотря на сравнительно небольшой выход фенола, полученный в этих работах, было ясно, что найдена перспективная основа для создания новых катализаторов. Дальнейшие исследования [64—74] позволили значительно улучшить каталитические свойства цеолитов и расширить круг катализируемых ими реакций, включая окисление закисью азота различных производных бензола.
В настоящее время на этой основе фирмой «Solutia» (бывшая фирма «Monsanto», США) и Институтом катализа СО РАН ведется создание нового фенольного процесса, который успешно прошел пилотные испытания и принят фирмой к промышленному освоению [75].Природа каталитической активности цеолитов Открытие цеолитных катализаторов, представляющих собой весьма нетипичные системы для окислительного катализа, поставило ряд вопросов фундаментального характера, среди которых вопрос о природе каталитической активности цеолитов вызывает наиболее оживленные дискуссии.
Для объяснения каталитической активности цеолитов были выдвинуты две гипотезы. Одна из них основана на традиционных представлениях о цеолитах как кислотных катализаторах, обладающих сильными бренстедовскими и льюисовскими кислотными центрами. Объяснение, исходящее из представлений о бренстедовских центрах, предполагает их способность протонировать молекулы закиси азота, что приводит к образованию гидроксил-катионов, которые далее атакуют молекулы бензола:
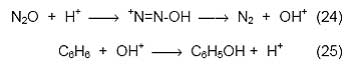
Эта гипотеза впервые была высказана в работе [61] и затем, с теми или иными оговорками, принята рядом других авторов [64, 66]. Однако специальное исследование, выполненное позднее с привлечением инфракрасной спектроскопии, не подтвердило эту гипотезу [76].
Ряд исследователей связывают активность цеолитов с льюисовскими кислотными центрами, которые образуют координационно ненасыщенные атомы алюминия или галлия, вышедшие из кристаллической решетки [65,67, 68, 71]. Это объяснение основывается на том, что с увеличением жесткости условий высокотемпературной обработки цеолита, проводимой на воздухе [65, 69], в вакууме [76] или в присутствии паров воды [71, 77], возрастает как концентрация льюисовских кислотных центров, так и каталитическая активность цеолита в реакции окисления бензола в фенол. Однако авторы этих работ не приводят каких-либо количественных корреляций между кислотностью и каталитической активностью.
В работах Института катализа СО РАН развивается другой подход, основанный на представлениях окислительного катализа. Исходная позиция этого подхода заключается в том, что сама по себе алюмосиликатная матрица цеолита, независимо от её кислотных свойств, неспособна катализировать такую тонкую и трудную реакцию как окисление бензола в фенол. Причина каталитической активности, вероятнее всего, связана с примесью какого-либо переходного металла. Действительно, как выяснилось в дальнейшем, таким металлом оказалось железо наиболее распространенная примесь, вносимая в основном с реагентами на стадии синтеза цеолита [60]. Роль железа была детально исследована на примере двух специально синтезированных Fe-содержащих цеолитных систем со структурой ZSM-5. Железо вводилось на стадии синтеза цеолитов и в одном случае его включали в Al-Si матрицу [78], а в другом – в чисто силикатную матрицу, синтезированную в отсутствие алюминия [79].
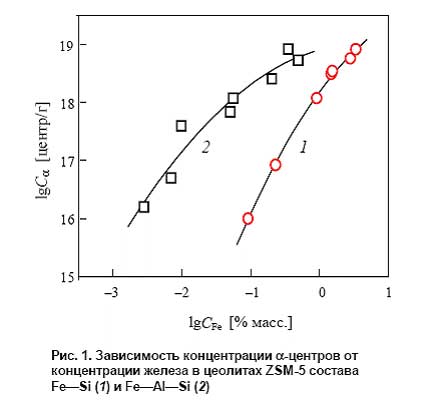
В обоих случаях самые чистые образцы, которые авторам удалось приготовить, принимая максимальные предосторожности против загрязнения же лезом (0,002-0,003 % масс. Fe), оказались неактивными. Активность появлялась только с введением железа, увеличиваясь по мере роста его концентрации. Это связано с тем, что железо в цеолите образует особые активные центры, названные α-центрами (их природа рассмотрена ниже), которые и обеспечивают протекание каталитической реакции. На рис. 1 показана зависимость концентрации каталитически активных центров от содержания Fe для обеих цеолитных систем.
Несмотря на сходный характер, кривые существенно сдвинуты относительно друг друга по оси абсцисс. Так, введение в алюмосиликатную матрицу железа уже на уровне сотых долей процента приводит к образованию значительного количества α-центров, тогда как в случае силикатной матрицы для этого требуется ввести в 10-30 раз большее количество железа.
Это говорит о том, что присутствие алюминия способствует образованию α-центров, хотя причина этого пока остается неясной. Одно из объяснений может заключаться в том, что активные комплексы Fe занимают катионно-обменные позиции в цеолите и стабилизируются на атомах алюминия [68]. Интересно, что независимо от состава матрицы и количества введенного железа α-центры проявляюудивительную равноценность в своих каталитических свойствах.
Это относится как к реакции разложения N2O [80], так и к реакции окисления бензола в фенол [60]. По отношению к последней реакции такой эффект хорошо виден из рис. 2, на котором приведена зависимость конверсии бензола от концентрации α-центров. Активность всех исследованных образцов описывается единой зависимостью и определяется только концентрацией α-центров.
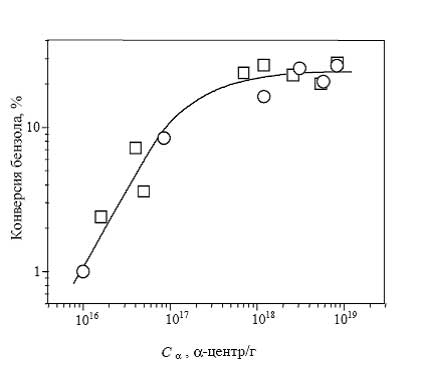
Рис. 2. Зависимость конверсии бензола от концентрации α-центров на цеолитах ZSM-5 состава Fe-Si (!) иFe-Al-Si (") при 350 °С
Согласно квантовохимическим расчетам [81], α-центры представляют собой биядерные комплексы железа, которые формируются на стадии выхода Fe из кристаллической решетки цеолита при его термических обработках. Аналогично внерешеточному алюминию, эти комплексы являются координационно -ненасыщенными частицами и обладают льюисовской кислотностью. Учитывая сходный механизм образования внерешеточных частиц Fe и Al, естественно ожидать некоторую корреляцию между каталитической активностью и льюисовской кислотностью, что и наблюдали при различных высокотемпературных обработках одного и того же образца цеолита. Однако в случае образцов с различным химическим составом такая корреляция не должна существовать, что действительно и подтверждается [79].
Это объясняется тем, что в разных образцах неодинакова концентрация внерешеточных атомов Al, которые дают основной вклад в льюисовскую кислотность, но не дают вклада в каталитическую активность. Квантовохимические расчеты также не подтверждают каталитическую роль льюисовских центров [82].Каталитическая активность железа в реакциях окисления закисью азота подтверждена в ряде работ других авторов [69, 70, 83].
Интересно, что образование α-центров приводит к качественному изменению в состоянии железа по сравнению с атомами Fe на поверхности оксида Fe2O3.
В составе α-центров атомы Fe теряют способность активировать молекулярный кислород, но приобретают повышенную способность к активации молекул N2O [80].
Это ярко проявляется в реакции окисления бензола с помощью этих молекул (табл. 2). Так, в присутствии закиси азота конверсия бензола составляет 27% при 350 оС, тогда как в присутствии О2 - только 0,3% при 500 оС. Более того, при этом изменяется направление реакции: если при действии N2O с высокой селективностью образуется фенол, то в случае О2 регистрируются лишь продукты полного окисления. На оксиде железа ни один из окислителей не приводит к образованию фенола.
ГЕННАДИЙ ИВАНОВИЧ ПАНОВ - доктор химических наук, заведующий лабораторией окислительного катализа на цеолитах Института катализа им. Г. К. Борескова СО РАН. Область научных интересов: окислительный катализ, механизм каталитических реакций.
АЛЕКСАНДР СЕРГЕЕВИЧ ХАРИТОНОВ - доктор химических наук, ведущий научный сотрудник Института катализа им. Г. К. Борескова СО РАН. Область научных интересов: каталитическое окисление органических соединений, цеолиты, технология каталитических процессов.
630090 Новосибирск, просп. акад. Лаврентьева, 5, Институт катализа СО РАН, тел. (3832)34-44-52, факс (3832)34-30-56, E-mail Panov@catalysis.nsk.su, Khar@catalysis.nsk.su
Подробнее с текущей ситуацией и прогнозом развития российского рынка фенола и ацетона можно познакомиться в отчете Академии Конъюнктуры Промышленных Рынков «Рынок фенола и ацетона в России».