Прогресс в области окислительного катализа (Часть II)
Представляем технологию окисления бензола в фенол закисью азота, разрабатываемую в Институте катализа им. Г. К. Борескова СО РАН.
Специфика действия N2O как окислителя

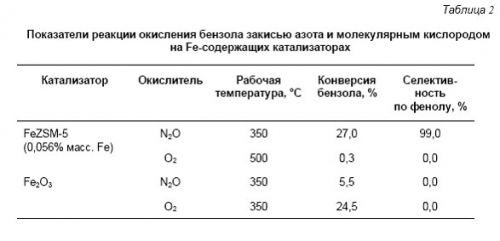
Специфика закиси азота как окислителя - это один из наиболее важных вопросов, возникающих при исследовании реакции окисления бензола в фенол. Преимущества закиси азота по сравнению с O2 наглядно демонстрируют результаты, представленные в табл. 2.
Естественно предположить, что столь сильное влияние природы окислителя связано с существенным изменением в состоянии поверхностного кислорода. Это предположение стимулировало постановку ряда работ по исследованию механизма реакции разложения закиси азота на цеолитах FeZSM-5, поскольку именно за счет этой реакции осуществляется поставка кислорода на поверхность катализатора.
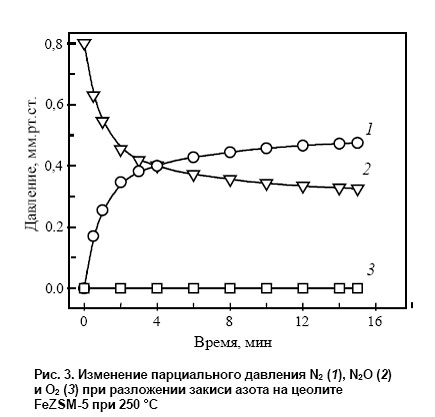
Исследования позволили установить, что разложение N2O протекает на α-центрах цеолита с образованием новой формы поверхностного кислорода, условно названной α-кислородом [84, 85]. В низкотемпературной области реакция образования α-кислорода носит стехиометрический характер:N2O + ( )α = (O)α + N2(26)
На рис. 3 приведена кинетическая кривая низкотемпературного разложения N2O в статической установке.
Видно, что протекание реакции сопровождается выделением в газовую фазу только N2, тогда как образующийся кислород заполняет α-центры и остается полностью связанным на поверхности. По мере заполнения α-центров реакция прекращается.
До температуры 300 оС α-кислород термически устойчив. Выше этой температуры он необратимо десорбируется в газовую фазу и реакция разложения N2O приобретает
характер обычного каталитического процесса.
Для проведения дальнейших количественных исследований важно знать концентрацию α-кислорода. Существует несколько методов, позволяющих надежно определить данную величину. Это может быть выполнено как путем непосредственного измерения количества выделившегося N2 (или израсходованного N2O) в ходе реакции (26) или путем десорбции Оα в газовую фазу при нагревании образца выше 300 оС. Кроме того, удобно использовать реакцию изотопного обмена, в которую α-кислород легко вступает уже при комнатной температуре:
(16O)α + 18O2 = (18O)α + 16O18O (27)
В состоянии равновесия доли изотопа 18О в поверхностном α-кислороде и в кислороде газовой фазы равны, что позволяет рассчитать количество Оα, вступившего в обмен. Собственный кислород цеолита инертен в отношении изотопного обмена и не мешает измерению концентрации α-кислорода.
Исходя из максимального количества α-кислорода, которое может быть «посажено» на данном образце цеолита (уравнение 26), рассчитывается концентрация α-центров Сα в предположении, что один атом кислорода занимает один центр. Значения Сα, определенные разными методами, хорошо согласуются между собой [85]. Для наиболее активных образцов с достаточно высоким содержанием железа величина Сα может достигать 100 мкмоль/г. Изучению свойств α-кислорода посвящено значительное число работ, результаты которых рассмотрены в обзоре [86]. Особенно следует отметить низкую энергию связи Оα с поверхностью и очень высокую реакционную способность, благодаря которой он способен уже при комнатной температуре окислять различные углеводороды.
В работах [81, 82, 87] предложена квантовохимическая модель активных центров, позволяющая описать особенности α-кислорода. Отметим, что α-кислород не образуется при адсорбции О2.Стехиометрическая реакция бензола с α-кислородом
Установление специфической способности закиси азота генерировать α-форму поверхностного кислорода подводит нас к главному вопросу относительно механизма реакции, а именно, к вопросу об участии α-кислорода в образовании фенола.
Согласно общепринятой точке зрения, которая особенно ясно сформулирована в работах школы Борескова применительно к классическим оксидным катализаторам [88], поверхностный кислород, участвующий в парциальном окислении, не должен обладать
низкой энергией связи с поверхностью и не должен иметь высокую реакционную способность, что находится в явном противоречии со свойствами α-кислорода.
Поэтому идея соотнести образование фенола с генерацией α-кислорода требует серьезного экспериментального подтверждения.
Следует отметить, что идентификация поверхностных форм кислорода, принимающих участие в реакциях окисления, представляет собой очень трудную задачу.
В условиях катализа при повышенных температурах происходят быстрые взаимные превращения различных форм кислорода [89, 90], что делает результаты малоинформативными. При пониженной температуре, когда таких превращений нет, идентификацию поверхностных форм кислорода обычно также не удается провести либо из-за их низкой активности, либо из-за малой концентрации. В случае же α-кислорода ситуация кажется уникальной- высокая активность и большая концентрация этой формы кислорода, которую можно регулировать (путем введения Fe) в пределах нескольких порядков. Это обстоятельство позволило нам простым и надежным способом ответить на вопрос об участии α-кислорода в реакции окисления бензола [85]
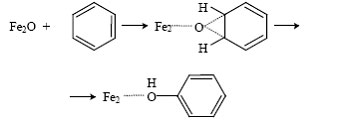
Сущность экспериментов заключалась в титровании α-кислорода бензолом, которое проводилось по схеме, включающей «посадку» кислорода на α-центр, его взаимодействие с бензолом при комнатной температуре и экстракцию продукта метанолом:
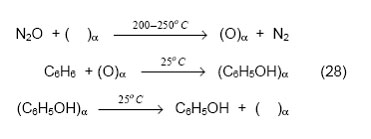
Как показали исследования, в пределах точности эксперимента выход фенола близок к теоретическому. Других продуктов реакции не обнаружено. Полученные результаты, несомненно, говорят об участии α-кислорода в образовании фенола. Позднее этот вывод был подтвержден дополнительными экспериментами с использованием α-кислорода, обогащенного изотопом 18О. Биомиметические свойства α-кислорода
Факт образования фенола при комнатной температуре в рассматриваемой реакции наводит на мысль о вероятном сходстве α-кислорода с активным кислородом монооксигеназ, для которых гидроксилирование ароматических соединений является типичной реакцией [17, 18, 91]. Напомним, что монооксигеназами называют ферменты, способные при комнатной температуре селективно вести окислительные реакции, в ходе которых один атом кислорода, переведенный в активное состояние, присоединяется к неактивированной молекуле углеводорода-субстрата, давая ОН-содержащее соединение, а второй - расходуется на образование воды.
Активация кислорода является наиболее трудной проблемой биомиметической химии, особенно в случае моделирования так называемых метанмонооксигеназ. Биядерные Fe-содержащие центры метанмонооксигеназ способны генерировать кислородные частицы, которые по активности значительно превосходят кислород других монооксигеназ. Вследствие этого, помимо способности гидроксилировать ароматические и другие соединения, метанмонооксигеназы обладают уникальной возможностью гидроксилировать даже метан наиболее инертную органическую молекулу.Изучение реакции α-кислорода с метаном [85, 92] с использованием той же трехстадийной схемы титрования, что и в вышеприведенном случае с бензолом («посадка» Oα, его взаимодействие с метаном при комнатной температуре и экстракция продукта) показало, что реакция протекает очень быстро, с образованием только метанола, количество которого соответствует количеству прореагировавшего метана.
Аналогичные результаты были получены для реакций с этаном, пропаном и рядом других углеводородов: окисление α-кислородом при комнатной температуре приводит к образованию тех же продуктов, что и при окислении с помощью монооксигеназ. Для установления более глубокой аналогии α-кислородного окисления с биологическим окислением важно сравнить не только продукты, но и механизм, по которому они образуются в обоих случаях.
Удобным инструментом для этой цели является измерение кинетического изотопного эффекта (КИЭФ). Известно, что биологическое окисление метана протекает с большим изотопным эффектом (от 5 до 12), тогда как в случае окисления бензола кинетический эффект не наблюдается [93]. Для сопоставления механизмов обсуждаемых процессов в работе [94] было проведено измерение КИЭФ для реакций окисления метана и бензола α-кислородом. Молекулы метана CH2D2, которые использовались в работе [94], могут реагировать либо по C-H, либо по C-D связи, давая две изотопные разновидности мета Экстрагированный с поверхности катализатора метанол был проанализирован методом ЯМР и на основании ЯМР-спектров рассчитаны величины изотопного эффекта. Оказалось, что в зависимости от температуры КИЭФ меняется от 5,5 при -50 оС до 1,9 при +100 оС. Высокие значения КИЭФ убедительно указывают на тот же механизм реакции, что и при окислении с помощью метанмонооксигеназ. В обоих случаях лимитирующая стадия реакции включает разрыв связи С-Н.
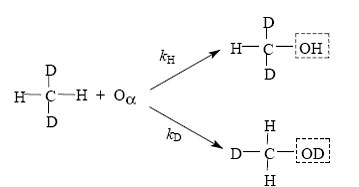
При взаимодействии α-кислорода с бензолом также образуются две изотопные разновидности фенола, содержащие ОН- и OD-группы. Результаты измерения КИЭФ и здесь показали сходство с биологическим процессом: как и в случае действия монооксигеназ, окисление бензола α-кислородом протекает без изотопного эффекта. Это значит, что лимитирующая стадия реакции не включает разрыв C—H связи и, вероятно, протекает через промежуточное образование ареноксида [94]:
Таким образом, α-кислород во многих отношениях аналогичен активному кислороду метанмонооксигеназ: он координирован на комплексах Fe, обладает высокой реакционной способностью и направляет окисление по механизму того же типа.
Активное состояние железа в цеолитной матрице При изучении активного состояния железа, входящего в состав α-центров, возникают два вопроса: о локализации α-центров и о структуре α-центров.
В общем случае железо в матрице цеолита может быть локализовано в трех состояниях [95]:
1) как изолированные ионы в тетраэдрических позициях кристаллической решетки (вместо Si);
2) как изолированные ионы или малые кластеры внутри микропористого пространства цеолита;
3) как частицы оксида на внешней поверхности кристаллов цеолита.
Было проведено несколько специальных исследований [96—99], чтобы понять, какому из этих состояний соответствуют α-центры. Результаты этих исследований позволили надежно исключить первое и третье состояния, так что α-центры, включающие Fe, могут находиться только во внутрикристаллическом пространстве цеолита.
Вопрос о структуре α-центров более сложный. Анализ результатов, проведенный в работе [100], указывает на то, что α-центры, вероятно, представляют собой биядерные комплексы железа. В пользу этого вывода свидетельствуют адсорбционные измерения, результаты ЭПР [80], квантовохимические расчеты [81, 87] и данные мессбауэровской спектроскопии [101, 102]. Отметим, что последний метод широко применяется для анализа Fe в составе метанмонооксигеназ. Использование мессбауэровской спектроскопии по отношению к цеолиту FeZSM-5 позволило обнаружить удивительное согласие между спектральными параметрами Fe в цеолите и в составе биядерных центров метанмонооксигеназ [100].
Косвенным доводом в пользу биядерного строения α-центров можно считать то обстоятельство, что биядерные оксо- (или гидроксо-)комплексы металлов, по-видимому, широко используются в природе для генерации высокоактивных частиц кислорода. Так, предполагается, что ферменты, осуществляющие окисление воды, а также их искусственные модели содержат биядерные комплексы в качестве функциональных единиц [103].Тем не менее, имеющихся экспериментальных данных пока недостаточно, чтобы считать биядерную структуру α-центров окончательно доказанной. Полностью нельзя исключить и моноядерный вариант структуры.
Как показали мессбауэровские измерения, «посадка» и снятие Oα сопровождается обратимыми окислительно-восстановительными переходами железа Fe2+ Fe3+ в составе FeZSM-5. Состояние Fe в матрице цеолита ZSM-5 является предметом изучения многих исследовательских групп [104—109]. В ряде работ также приводятся аргументы в пользу образования биядерных комплексов железа [104, 105, 107]. Однако следует отметить, что в этих работах, как правило, имеют дело с более высокими концентрациями Fe. В настоящее время трудно сказать, в какой мере комплексы, наблюдаемые этими исследователями, соотносятся с α-центрами, обсуждаемыми в данной работе.
Новый фенольный процесс. Технологические аспекты Разработка нового фенольного процесса, основанного на прямом окислении бензола закисью азота, ведется в рамках сотрудничества между Институтом катализа СО РАН и американской фирмой «Solutia», которая является новой химической компанией, отделившейся от фирмы «Monsanto» около двух лет назад. «Solutia»принадлежит к числу крупнейших в мире производителей адипиновой кислоты (полупродукта для синтезанайлона 6,6), что в значительной мере и определило её первоначальный интерес к каталитической химии закиси азота.
Технологическая схема получения адипиновой кислоты (рис. 4) включает следующие стадии: гидрирование бензола в циклогексан, окисление циклогексана кислородом воздуха в смесь циклогексанола и циклогексанона и, наконец, дальнейшее окисление этой смеси в адипиновую кислоту с помощью азотной кислоты. Последняя стадия дает большое количество отходов закиси азота, которая образуется приблизительно в мольном соотношении 1:1 с адипиновой кислотой. В настоящее время закись азота уже не рассматривается как безобидный «веселящий газ». Она обладает сильным парниковым эффектом [110], который (в расчете на моль) в 160 раз превосходит эффект СО2, главного «парникового газа». Кроме того, имея большое время жизни, N2O достигает верхних слоев атмосферы, где способствует разрушению озонового слоя Земли. Если учесть, что человек ежегодно выбрасывает в атмосферу более 10 млн. тонн N2O (из них около 10% связано с производством адипиновой кислоты), то можно понять, почему закись азота вызывает растущее беспокойство экологов и почему в ряде стран принимаются законы, ограничивающие выбросы N2O.
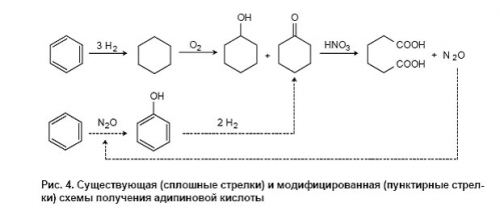
В этой ситуации фирма «Solutia» предложила идею использовать отходы закиси азота для окисления бензола в фенол и включить эту реакцию как ключевую стадию в модифицированную схему адипиновой кислоты (см. рис. 4). По этой схеме бензол сначала окисляется в фенол с помощью N2O и лишь затем идет на стадию гидрирования. Закись азота, образующаяся на последней стадии, возвращается в начало процесса, замыкая, таким образом, свой технологический цикл. Новый фенольный процесс основан на использовании Fe-содержащих цеолитов ZSM-5, и по имени α-кислорода назван AlphOx-процессом. Окисление бензола осуществляется в простом адиабатическом реакторе, что имеет немаловажное значение с точки зрения экономичности процесса. В табл. 3 приведены типичные технологические показатели процесса на пилотном заводе, построенном фирмой в г. Пенсакола (США) [111].
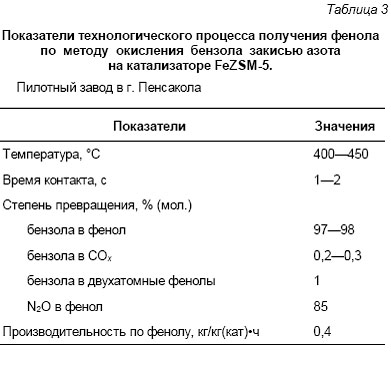
Производство функционирует в режиме непрерывной рециркуляции газовых и жидких потоков с выделением из продуктов чистого фенола. Процесс обладает высокой селективностью в расчете как на бензол, так и назакись азота. Его производительность (400 г фенола с 1кг катализатора в час) является высоким показателем для реакций селективного окисления. Кроме того,AlphOx-процесс обладает рядом других преимуществ по сравнению с кумольным методом, а именно: одностадийность, отсутствие взрывоопасных промежуточных веществ, высокая безопасность и экологичность процесса [111]. Таким образом, вместо траты средств на нейтрализацию закиси азота новый процесс использует её как ценное химическое сырье.
По результатам пилотных испытаний фирмой принято решение о строительстве крупного промышленного завода мощностью 140 тыс. тонн фенола в год. Как показывают оценки, новый процесс будет выгодным и в случае специального производства закиси азота, которая может быть с высокой селективностью получена путем окисления аммиака молекулярным кислородом [112]. С этой целью между Институтом катализа и фирмой «Solutia» заключен дополнительный договор на разработку крупномасштабного процесса
получения закиси азота.
Заключение
Окислительный катализ является одной из самых динамичных и плодотворных областей каталитической химии, развитие которой безусловно находится на подъеме. Отмечая большие успехи, достигнутые в этой области в течение последних четырех десятилетий,
вместе с тем мы должны признать, что две труднейшие проблемы окислительного катализа, а именно, прямое гидроксилирование бензола молекулярным кислородом, так же как и прямое гидроксилирование метана, по-прежнему остаются вызовом, который не нашел ответа и который мы передаем новому поколению исследователей и новому веку.
Решению этих проблем, вероятно, во многом будетспособствовать прогресс в понимании механизма действия природных ферментов-монооксигеназ, обладающих удивительной способностью проводить эти реакции в обычных условиях. Наше краткое знакомство с биомиметическим подходом говорит о том, что разрыв,который обычно подразумевается между биологическим катализом, с одной стороны, и химическим катализом, с другой, на самом деле не так велик, как может показаться на первый взгляд. Здесь открывается широкий простор для взаимного обогащения и сотрудничества.
Объединение опыта и таланта исследователей, работающих в этих, пока разъединенных, областях катализа несомненно приведет к новым открытиям и новым успехам.
Подробнее с текущей ситуацией и прогнозом развития российского рынка фенола и ацетона можно познакомиться в отчете Академии Конъюнктуры Промышленных Рынков «Рынок фенола и ацетона в России».
.
ЛИТЕРАТУРА
1. Справочник химика. М.-Л.: Госхимиздат, 1963, т. 1, с. 22.
2. Haber J. In: Perspectives in Catalysis. Eds. J. M. Thomas, K.I.
Zamaraev, Oxford. Blackwell Scientific Publications, 1992,p. 371.
3. Maulijn J.A., Van Leeuwen P.W.N.M., Van Santen R.A. Stud. Surf. Sci. Catal., 1993, v. 79, p.3.
4. Sheldon R.A., Dakka J. Catal. Today, 1994, v. 19, p. 215.
5. Cavany F., Trifiro F. Ibid., 1997, v. 34, p. 269.
6. Моисеев И.И., Варгафтик М.Н., Сыркин Ю.К. Докл. АН СССР, 1960, т. 130, с. 820; т. 133, с. 377.
7. Крылов О.В., Марголис Л.Я. Проб. кинетики и катализа. М.: Наука, 1985, с. 5.
8. Sokolovsky V.D. Catal. Revs Sci. and Eng., 1990, v. 32, p. 1.
9. Andrushkevich T.V. Ibid., 1993, v. 35, № 2, p. 213.
10. Krylov O.V. Catal. Today, 1993, v. 18, p. 209.
11. Haber J. Stud. Surf. Sci. Catal., 1997, v. 110, p 1.
12. Замараев К.И. Химия в интересах устойчивого развития, 1993, т. 1, № 1, с. 149.
13. Hьlderich W.F. New Frontiers in Catalysis, Amsterdam: Elsevier,1993, p. 127.
14. Heck R.M., Farrauto R.J. Catalytic Air Pollution. N.Y.: Van Nostrand Reinhold, 1995.
15. Parmon V.N., Fibutch H., Bridgwater A., Hall D.O. Chemistry for the Energy Future. IUPAC XXI century monograph. Oxford: Blackwell, 1999.
16. Roth J.F. Chem. Eng. News, 1993, v. 31, p. 27.
17. Dalton H. Catal. Today, 1992, v. 13, p. 445.
18. Шилов А.Е. Активация и каталитические реакции алканов. М.:Мир, 1992, с. 12.
19. Метелица Д.И. Активация кислорода ферментативными системами. М.: Наука, 1982.
20. Кружалов Б.Д., Голованенко Б.И. Совместное получение фенола и ацетона. М.: Наука, 1963, 200 с.
21. Kirk-Othmer. Encyclopedia of Chemical Technology. 3rd Edition, v.17, p. 373, see article «Phenol».
22. Brownstain A.M. CHEMTECH, September, 1994, p. 58.
23. Miki J., Asanuma M., Tashibana Y., Shikoda T. Bull. Chem. Soc. Japn., 1995, v. 68, p. 2429.
24. ChemExpo, Phenol (1999, March 29) http://www.chemexpo.com/news/PROFILE990329.cfm
25. Полищук В. Химия и жизнь, 1988, № 8, с. 68.
26. Авт. свид. СССР № 7238 и № 7240 от 7.01.1947.
27. Толстиков Г.А. Панорама нефтехимии, 1997, № 4, с. 76.
28. Харлампович Г.Д., Чуркин Ю.В. Фенолы. М.: Химия, 1974, 376с.
29. SRI report No. 22C, 2-2 (Appl. Catal. A, 1994, v. 114, № 1, p. N2).
30. Марек Л.Ф., Ган Д.А. Каталитическое окисление органических соединений. М.: ОНТИ, 1936, с. 446.
31. Голодец Г.И. Гетерогенно-каталитическое окисление органических веществ. Киев: Наукова думка, 1978, с. 209.
32. Sheldon R.A. Top. Curr. Chem., 1993, v. 164, p. 21.
33. Sasaki K., Kitano T., Nakai T. e. a. New Developments in Selective Oxidation II, Elsevier, 1994, p. 451.
34. Zakharov V.Yu., Zakharova O.M., Romanovsky B. V., Mardaleishvili R.E. React. Kinet. and Catal. Lett., 1977, v. 6, p. 133.
35. Sheldon R.A., Arends I.W.C.E., Lempers H.E.B. Catal. Today, 1998, v. 41, p. 387.
36. Караханов Э.А., Волков С.М., Дедов А.Г. Катализ. М.: изд. МГУ, 1987, с. 147.
37. Notary B. Adv. Catal., 1996, v. 41, p. 253.
38. Belussi G., Rigutto M.S. Stud. Surf. Sci. Catal., 1994, v. 85, p. 177.
39. De Vos D.E., Buskens P.L., Vanoppen D.L. e.a. Compr.Supramol. Chem., 1996, v. 7, p. 647.
40. Arends I.W.C.E., Sheldon R.A., Wallau M., Schuchardt U. Angew. Chem., 1997, Bd. 36, S. 1144.
41. Заявка 62-67038, Япония. Заявл. 20.09.85, № 60-206234,опубл. 26.03.87. МКИ С 07 С 37/60, С 07 С 39/04.
42. Sittig M. Organic Chemical Process Encyclopedia. 2nd ed., 1969, p.517.
43. Mori M., Nakai T., Jahiro H., Nata M., Sasaki K. Bull. Chem. Soc.Jap., 1995, v. 68, № 6, p. 1747.
44. Shengchun CaO e. a. Chin. J. Petrochem. Tech., 1995, v. 10, p.708 [Chem. Abstracts, 1995, v. 123].
45. Moro-oka Y., Akita M. Catal. Today, 1998, v. 41, p. 327.
46. Kuznetsova N.I., Detusheva L.G., Kuznetsova L.I. e.a. J. Mol.Catal. A: Chemical, 1996, v. 114, p. 131.
47. Шилов Н.А. О сопряженных реакциях окисления. М., 1905, 304 с.; Нагиев Т.М. Химическое сопряжение. М.: Наука, 1989, 216 с.
48. Clerici M. G., Ingallina P. Catal. Today, 1998, v. 41, p. 351.
49. Kitano T., Wani T., Ohnishi T. e.a. Catal. Lett., 1991, v. 11, p. 11.
50. Jintoku T., Nishimura K., Takaki K., Fujiwara Y. Chem. Lett., 1991, v. 193.
51. Иоффе И.И., Левин Я.С., Кронич И.Г. Ж. физ. химии, 1959, т.33, № 4, с. 863.
52. Sasaki K., Ito S., Sahek Y., Kinoshita T., Yamasaki T., Harada J. Chem. Lett., 1983, v. 37.
53. Kitajima N., Ito M., Fukui H., Moro-oka Y. J. Chem. Soc. Chem. Communs., 1991, p. 102.
54. Vayenas C.G., Bebelis S.I. Stud. Surf. Sci. Catal., 1997, v. 110, p.77.
55. Galvita V.V., Belyaev V.D., Demin A.K., Sobyanin V.A. Appl. Catal. A: General, 1997, v. 165, p. 301.
56. Otsuka K., Yamanaka I. Catal. Today, 1998, v. 41, p. 311.
57. Iwamoto M., Matsukami K., Kagawa S. J. Phys. Chem., 1983, v.
ГЕННАДИЙ ИВАНОВИЧ ПАНОВ - доктор химических наук, заведующий лабораторией окислительного катализа на цеолитах Института катализа им. Г. К. Борескова СО РАН. Область научных интересов: окислительный катализ, механизм каталитических реакций.
АЛЕКСАНДР СЕРГЕЕВИЧ ХАРИТОНОВ - доктор химических наук, ведущий научный сотрудник Института катализа им. Г. К. Борескова СО РАН. Область научных интересов: каталитическое окисление органических соединений, цеолиты, технология каталитических процессов.
630090 Новосибирск, просп. акад. Лаврентьева, 5, Институт катализа СО РАН,
тел. (3832)34-44-52,
факс (3832)34-30-56,